

A Comprehensive Review of Small-Molecule Inhibitors Targeting Bruton Tyrosine Kinase: Synthetic Approaches and Clinical Applications
1
Nanyang Central Hospital, Nanyang 473000, China
2
First People’s Hospital of Shangqiu, Shangqiu 476100, China
3
The Rogel Cancer Center, Department of Internal Medicine, University of Michigan, Ann Arbor, MI 48109, USA
4
Department of Orthopedics, China-Japan Union Hospital, Jilin University, Changchun 130033, China
*
Authors to whom correspondence should be addressed.
Molecules 2023, 28(24), 8037; https://doi.org/10.3390/molecules28248037
Submission received: 1 November 2023
/
Revised: 7 December 2023
/
Accepted: 8 December 2023
/
Published: 11 December 2023
(This article belongs to the Special Issue Synthesis and Evaluation of Bioactivity of Enzyme Inhibitors)
Abstract
:Bruton tyrosine kinase (BTK) is an essential enzyme in the signaling pathway of the B-cell receptor (BCR) and is vital for the growth and activation of B-cells. Dysfunction of BTK has been linked to different types of B-cell cancers, autoimmune conditions, and inflammatory ailments. Therefore, focusing on BTK has become a hopeful approach in the field of therapeutics. Small-molecule inhibitors of BTK have been developed to selectively inhibit its activity and disrupt B-cell signaling pathways. These inhibitors bind to the active site of BTK and prevent its phosphorylation, leading to the inhibition of downstream signaling cascades. Regulatory authorities have granted approval to treat B-cell malignancies, such as chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), with multiple small-molecule BTK inhibitors. This review offers a comprehensive analysis of the synthesis and clinical application of conventional small-molecule BTK inhibitors at various clinical stages, as well as presents promising prospects for the advancement of new small-molecule BTK inhibitors.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
BTK plays a crucial role as an essential enzyme in the signaling pathway of BCR activation [1,2,3]. B-cells, a type of white blood cell, play a crucial role in producing antibodies and depend on them for their growth and functioning. The abnormal activity of BTK has been linked to different B-cell cancers, autoimmune conditions, and inflammatory disorders [4,5].
At present, the utilization of small-molecule inhibitors targeting BTK has become a very promising treatment method for these diseases. These inhibitors work by binding to the active site of BTK and blocking its enzymatic activity, thereby inhibiting downstream signaling pathways that promote cell proliferation, survival, and inflammation [6]. Ibrutinib, the initial BTK inhibitor approved by the U.S. Food and Drug Administration (FDA), has demonstrated exceptional effectiveness in addressing B-cell malignancies like CLL and MCL [7]. However, Ibrutinib, the first-generation BTK inhibitor, still possesses certain limitations. It possesses the ability to inhibit various targets, including epidermal growth factor receptor (EGFR), tyrosine kinase expressed in hepatocellular carcinoma (TEC), bone marrow kinase on chromosome X (BMX), and others [8]. In order to solve these difficulties, many enterprises have begun to develop the second generation of BTK inhibitors [9,10]. Approved in 2017 and 2019, respectively, Acalabrutinib and Zanubrutinib have been structurally optimized and improved to have less off-target effects and greater effectiveness than Ibrutinib [11]. Since then, several other small-molecule BTK inhibitors have been created and are currently undergoing assessment in clinical trials for various indications. In the last ten years, a multitude of preclinical and clinical trials have examined the efficacy of BTK inhibitors as standalone treatments or in conjunction with targeted therapies, immunotherapy, or traditional chemotherapy to treat different types of cancers. In addition, as covalent BTK inhibitors still have off-target effects and tolerability problems, the industry and academia are increasingly calling for non-covalent BTK inhibitors. A new generation of BTK inhibitors, such as Pirtobrutinib, is in the fast lane of development [12,13].
The progress in the realm of BTK inhibitor research heralds a promising spectrum of therapeutic avenues for the effective management of afflictions characterized by BTK overexpression. Additional investigation and inquiry are essential to achieving a comprehensive understanding of the full potential inherent in these inhibitors. Further scrutiny and inquiry are paramount to attaining a comprehensive grasp of the inherent potential embodied by these inhibitors. As far as our current knowledge extends, a thorough investigation into the synthetic methodologies employed in chemical compounds and their respective mechanisms of action within clinical contexts holds substantial promise for propelling the advancement of groundbreaking pharmaceuticals.
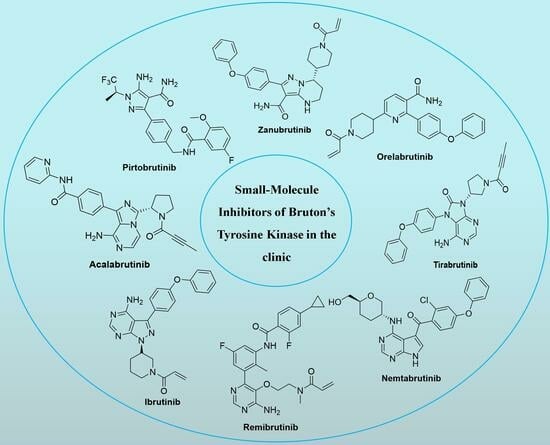
Drawing upon well-substantiated data concerning BTK inhibitors, this review methodically elucidates the clinical utility and synthetic techniques associated with prototypical BTK inhibitors across diverse clinical phases (Figure S1 and Table S1). This information carries significant import in the ongoing design and refinement of BTK inhibitors.
2. Signaling Pathway of BTK
BTK is a non-receptor tyrosine kinase that plays a crucial role in the signaling pathways of B-cells, macrophages, and microglia [14]. As shown in Figure 1, BTK is activated downstream of the BCR and FcγRIII in macrophages and microglia, leading to the activation of downstream elements crucial to immune cell function. When BTK is activated, it phosphorylates phospholipase Cγ2 (PLCγ2), causing the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds to its receptor on the endoplasmic reticulum, leading to the release of calcium ions into the cytoplasm, which activates downstream signaling pathways. DAG triggers the activation of protein kinase C (PKC), which subsequently initiates the activation of signaling pathways downstream. BTK also activates the nuclear factor kappa B (NF-κB) pathway, which is involved in the regulation of immune responses. BTK also participates in signaling networks that play a role in innate immunity, such as the transmission of signals through the Fc epsilon receptor (FcεR) in mast cells and basophils. When antigen binds to immunoglobulin E (IgE) molecules linked to FcεR, it triggers the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in the FcεR β and γ chains. During the BCR signal transduction process, the phosphorylation of FcεR and ITAMs plays a crucial role in recruiting and subsequently activating Lck/yes-related novel protein tyrosine kinase (LYN) and spleen tyrosine kinase (SYK).
3. Representative Small-Molecule BTK Inhibitors in the Clinic
3.1. Aminopyrimidines
3.1.1. Spebrutinib (CC-292)
Spebrutinib, developed by Xinji Company, is a small-molecule drug currently in clinical phase II. It is being investigated for its potential in treating diffuse large B-cell lymphoma (DLBCL) and CLL. Spebrutinib irreversibly binds to the cysteine residue (Cys481) in the active site of BTK, inhibiting its activity and downstream signaling pathways (IC50 = 0.5 nM) [15,16]. In clinical trials, Spebrutinib has shown favorable efficacy in patients with relapsed or refractory CLL and MCL [17]. It has demonstrated durable responses and manageable toxicity profiles, leading to its potential as a targeted therapy for these diseases. However, additional clinical trials are required to determine its effectiveness and safety in larger groups of patients [17].
In 2008, a method for synthesizing Spebrutinib was elucidated (Scheme 1) [18,19]. First, the 4-Cl of 2,4-dichloro-5-fluoropyrimidine (SPEB-001) is substituted with tert-butyl (3-aminophenyl)carbamate (SPEB-002) in the presence of N,N-diisopropylethylamine (DIPEA) to obtain SPEB-003. SPEB-003 undergoes further substitution with 4-(2-methoxyethoxy)aniline (SPEB-004) to obtain SPEB-005. Treatment of SPEB-005 with trifluoroacetic acid (TFA) to remove the t-butyloxycarbonyl (Boc) group gives SPEB-006. Finally, in the presence of DIPEA, SPEB-006 is amidated with acryloyl chloride to obtain Spebrutinib.
3.1.2. Evobrutinib (M-2951)
Evobrutinib, developed by Merck Serrano, is a small-molecule medication currently in the advanced clinical phase III of development. It is specifically designed to address the treatment of multiple sclerosis (MS). In clinical trials, Evobrutinib has shown efficacy in treating relapsing forms of MS. Phase 2 trials showed a notable decrease in the occurrence of fresh brain lesions and relapse rates when compared to the placebo group [20,21]. Additionally, Evobrutinib has shown potential to treat autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) [22]. Although additional research is required to comprehensively comprehend its long-term safety profile, Evobrutinib demonstrates promise as a valuable therapeutic choice for individuals suffering from autoimmune disorders.
The preparation of Evobrutinib first consists of the reaction of 5,6-dichloropyrimidin-4-amine (EVOB-001) with N-Boc piperidine EVOB-002 to give EVOB-003 (Scheme 2) [23]. Subsequently, Suzuki coupling of EVOB-003 with (4-phenoxyphenyl)boronic acid (EVOB-004) catalyzed by Pd(OAc)2 affords EVOB-005. Treatment of EVOB-005 with HCl to remove the Boc moiety gives EVOB-006. Finally, EVOB-006 is amidated with acryloyl chloride to obtain Evobrutinib.
3.1.3. Remibrutinib (LOU-064)
Remibrutinib is a small-molecule drug developed by Novartis pharmaceuticals. At present, the highest research and development stage of the drug is clinical phase III, which is used to treat chronic urticaria (CU), MS, urticaria, and relapsing remitting MS. Remibrutinib is a highly effective BTK inhibitor that can be taken orally. It has a remarkable potency, with an IC50 value of 1 nM. In blood, Remibrutinib demonstrates strong inhibition of BTK activity, with an IC50 value of 0.023 μM [24]. Remibrutinib effectively suppressed HuMOG experimental autoimmune encephalomyelitis (EAE), a B-cell-dependent disease, in a dose-dependent manner and significantly alleviated neurological symptoms. When administered orally at a dose of 30 mg/kg, Remibrutinib exhibited potent inhibition of BTK in both peripheral immune organs and the brain of EAE mice. In clinical trials, Remibrutinib has shown promising efficacy in patients with CLL and other B-cell malignancies. It has shown impressive response rates and long-lasting remissions in patients who have not responded to previous treatments. Additionally, Remibrutinib has shown a favorable safety profile with manageable toxicities, including mild to moderate gastrointestinal symptoms and reversible hematological abnormalities [25,26].
Scheme 3 presents a concise method for synthesizing Remibrutinib [24]. First, in the presence of ammonia, 4,6-dichloro-5-methoxypyrimidine (REMI-001) undergoes halogen amination to give the amine REMI-002, and then boron tribromide is used to remove the methyl group of REMI-002 to give the phenol REMI-003. In the presence of diisopropyl azodicarboxylate (DIAD) with Smopex-301, a convenient polymer-supported version of triphenylphosphine, REMI-003 reacts with hydroxyethylamine REMI-004 in a Mitsunobu reaction to give REMI-005. Under the catalytic effect of PdCl2(PPh3)2, REMI-005 reacts with the intermediate REMI-006 in a Suzuki coupling reaction to give REMI-007. The Boc group of REMI-007 is removed using TFA to obtain REMI-008. Finally, REMI-008 is amidated with acrylic acid to obtain Remibrutinib in the presence of the condenser propylphosphonic anhydride (T3P).
3.2. Pyrimidine-Fused Bicyclic Heterocycles
3.2.1. Ibrutinib (Imbruvica, PCI-32765)
Ibrutinib, originally created by Pharmacyclics LLC. and marketed as Imbruvica, received its first FDA approval to treat MCL on 13 November 2013. Subsequently, it obtained FDA approval for the management of Waldenstrom macroglobulinemia (WM), CLL, marginal zone B-cell lymphoma (MZBL), and graft vs. host disease (GvHD) in subsequent years. Ibrutinib is a BTK inhibitor that is both selective and irreversible, with an IC50 value of 0.5 nM [27]. Ibrutinib effectively blocks the activity of the enzyme by creating a covalent bond with Cys481, found in the active site of BTK. This blocking action of Ibrutinib on BTK then stops the phosphorylation process of downstream substrates like phospholipase C-γ (PLC-γ) [28]. The increase in IC50 against BTK-C481S phosphorylation from 2.2 nM to 1 μM is due to the fact that Ibrutinib cannot establish a covalent bond with the serine hydroxyl group [29].
An optimized synthetic route for Ibrutinib is described in Scheme 4 [30]. First, 4-phenoxybenzoyl chloride (IBRU-001) undergoes a Knoevenagel-type reaction with malony-dinitril to obtain the enol intermediate, followed by the methylation of the enol intermediate with dimethyl sulfate to obtain IBRU-002. In the presence of triethylamine (TEA), IBRU-002 undergoes pyrazole cyclization with hydrazine IBRU-003 to give IBRU-004. Finally, catalyzed by acetic acid, IBRU-004 undergoes a pyrimidine cyclization with the cycloadduct dimethylformamide dimethylacetal (DMF-DMA) to obtain Ibrutinib.
3.2.2. Zanubrutinib (Brukinsa, BGB-3111)
Zanubrutinib, developed by BeiGene, received its initial FDA approval on 14 November 2019, to treat MCL. Subsequently, it gained approval to treat MZBL, CLL, and WM. Zanubrutinib, a member of the second-generation BTK inhibitor drug category, demonstrates exceptional effectiveness and selectivity towards BTK while causing minimal off-target effects in comparison [31]. Due to its similar binding specificity to other BTK inhibitors, Zanubrutinib impedes BTK function by forming a covalent bond with Cys481. Due to this binding pattern, Zanubrutinib has the ability to bind to adenosine triphosphate (ATP)-binding kinases that possess a Cys481 at this specific site, regardless of their relationship or similarity, exhibiting varying levels of affinity [31,32,33].
Zanubrutinib is prepared by the reaction of 4-phenoxybenzoic acid (ZANU-001) with thionyl chloride to obtain the chloride intermediate IBRU-001, and then IBRU-001 undergoes a Knoevenagel-type reaction with malony-dinitril to obtain ZANU-002 in the presence of DIPEA (Scheme 5) [34]. Alkylation of ZANU-002 with trimethyl orthoformate affords IBRU-002, followed by cyclization of IBRU-002 with hydrazine hydrate to give pyrazol ZANU-003. Under the catalytic effect of acetic acid, ZANU-003 reacts with N-Boc piperidine ZANU-004 to give ZANU-005. Reduction of ZANU-005 using sodium borohydride gives ZANU-006, followed by hydrolysis of the cyano group of ZANU-006 using hydrogen peroxide to give the amide ZANU-007. Treatment of ZANU-007 with TFA to remove the Boc group yields the trifluoroacetate salt ZANU-008. Finally, ZANU-008 is first amidated with acryloyl chloride to obtain the amide ZANU-009, and then ZANU-009 is separated using Chiral pre-high-performance liquid chromatography (HPLC) to obtain Zanubrutinib.
3.2.3. Tirabrutinib (Velexbru, ONO/GS-4059)
Tirabrutinib Hydrochloride, created by Ono Pharmaceutical, was granted approval by the Pharmaceuticals and Medical Devices Agency (PMDA) on 25 March 2020. This drug is used for treating lymphoma [35]. Tirabrutinib, an orally administered BTK inhibitor (IC50 = 6.8 nM), possesses the capability to cross the blood–brain barrier (BBB) [36]. As a novel BTK inhibitor, Tirabrutinib exerts its anti-tumor and anti-inflammatory effects through selective inhibition of BTK activity. For example, Tirabrutinib has shown good efficacy in patients with CLL [37,38]. Common adverse events (AEs) were rash (35.3%) and vomiting (29.4%) [38].
Tirabrutinib Hydrochloride is prepared first by the reaction of dibenzylamine with nitropyrimidine TIRA-001 to give TIRA-002 (Scheme 6) [39]. In the presence of TEA, the reaction of TIRA-002 with aminopyrrolidine TIRA-003 gives amine TIRA-004. Reduction of TIRA-004 using zinc powder affords the amine TIRA-005, which is subsequently amidated with N,N′-carbonyl diimidazole (CDI) to give imidazolidinone TIRA-006. Under the 20% Pd(OH)2/C reduction, TIRA-007 is derived from TIRA-006 by removing the benzyl (Bn) group. A Chan–Lam coupling reaction of amide TIRA-007 with boronic acid TIRA-008 catalyzed by copper acetate gives TIRA-009, which is treated with hydrochloric acid to eliminate the Boc-protecting group, resulting in the formation of TIRA-010. In the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and hydroxybenzotriazole (HOBt), TIRA-010 is amidated with 2-butynoic acid (TIRA-011) to obtain TIRA-012. Finally, treatment of TIRA-012 with hydrochloric acid gives Tirabrutinib Hydrochloride.
3.2.4. Nemtabrutinib (MK-1026)
Nemtabrutinib was developed by Arqule Inc. At present, the highest research and development stage of the drug is clinical phase III, which is used to treat chronic lymphoblastic leukemia. Nemtabrutinib is a promising BTK inhibitor with potent activity against B-cell malignancies, including CLL and non-Hodgkin lymphoma (NHL) [40]. It has also shown promising activity against drug-resistant forms of these cancers. In clinical trials, Nemtabrutinib has shown favorable efficacy and safety profiles [41]. Nemtabrutinib has exhibited substantial clinical efficacy in patients with relapsed or refractory CLL and NHL, resulting in long-lasting responses. Moreover, it has displayed promise when used in combination with other targeted agents, thereby augmenting its effectiveness [42].
The approach for Nemtabrutinib is described in Scheme 7 [43]. First, pyrrolo[2,3-d]pyrimidine NEMT-001 undergoes lithium-bromine exchange with a lithium reagent to obtain an organolithium reagent, which is then added to methyl 2-chloro-4-phenoxybenzoate (NEMT-002) to obtain ketone NEMT-003. Finally, in the presence of DIPEA, the Cl of NEMT-003 is substituted with the amine NEMT-004 to obtain Nemtabrutinib.
3.2.5. Rilzabrutinib (PRN-1008)
Rilzabrutinib, created by Principia Biopharma Inc., is currently in the advanced clinical phase III of development. Its primary purpose is to treat idiopathic thrombocytopenic purpura (ITP). Rilzabrutinib, an orally active inhibitor of BTK (IC50 = 1.3 nM), is a reversible, covalent, and selective compound [44,45]. Rilzabrutinib has demonstrated encouraging effectiveness in the management of autoimmune disorders during clinical trials [46]. In a phase II trial involving patients with active RA, Rilzabrutinib significantly improved disease activity compared to placebo. Additionally, Rilzabrutinib has shown efficacy in the treatment of ITP and pemphigus vulgaris, with positive results observed in phase 2 trials [47].
The preparation of Rilzabrutinib is depicted in Scheme 8 [48]. First, the amine RILZ-001 is iodinated using N-iodosuccinimide (NIS) to obtain RILZ-002. In the presence of triphenylphosphine and DIAD, RILZ-002 is mixed with tert-butyl (R)-3-hydroxypiperidine-1-carboxylate (RILZ-003) in a Mitsunobu reaction to give RILZ-004. Catalyzed by Pd(dppf)Cl2, RILZ-004 undergoes a Suzuki coupling reaction with the intermediate RILZ-005 to give RILZ-006. RILZ-006 is treated with TFA to remove the Boc group to give RILZ-007. Subsequently, RILZ-007 is amidated with 2-cyanoacetic acid in the presence of the condenser 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) to give RILZ-008. Finally, RILZ-008 undergoes a Knoevenagel condensation reaction with the aldehyde RILZ-009 to obtain Rilzabrutinib.
3.2.6. Abivertinib (AC-0010)
Abivertinib, developed by Zhejiang ACEA Pharmaceutical Co., Ltd., has applied for listing to treat non-small cell lung cancer (NSCLC) in China. Abivertinib is a specific inhibitor of tyrosine kinases that targets mutant forms of human EGFR and BTK [49]. Abivertinib has been investigated for its potential use in the management of NSCLC and B-cell malignancies. It exerts its effects by binding to and inhibiting EGFR and BTK receptors, leading to immunomodulatory actions. These actions include the inhibition of pro-inflammatory cytokine production and release, such as tumor necrosis factor (TNF)-α and interleukins [50]. It has also shown favorable pharmacokinetic properties, with good oral bioavailability and tissue distribution. Nevertheless, additional research is required to thoroughly assess its long-term effectiveness and safety.
The preparation of Abivertinib starts with the N-alkylation of pyrrolo[2,3-d]pyrimidine ABIV-001 with 2-(trimethylsilyl)ethoxymethyl chloride (SEMCl) in the presence of sodium hydride to yield ABIV-002 (Scheme 9) [51]. Subsequently, in the presence of potassium carbonate, ABIV-002 undergoes a Williamson synthesis with 3-nitrophenol (ABIV-003) to give the ether ABIV-004. The Buchwald–Hartwig cross-coupling of ABIV-004 with aniline ABIV-005 catalyzed by Pd2dba3 affords ABIV-006, which is reduced with iron powder to give the amine ABIV-007. Under the action of DIPEA, ABIV-007 is amidated with acryloyl chloride to obtain ABIV-008. Finally, ABIV-008 is subjected to incomplete removal of the SEM group by TFA to obtain the hydroxymethyl intermediate ABIV-009, and then ammonia treatment is applied to remove the hydroxymethyl group to obtain Abivertinib.
3.3. Benzopyrroles
3.3.1. Luxeptinib (CG-806)
Luxeptinib, co-developed by Crystalgenomics Inc. and Aptose, is investigated for its potential in treating various conditions, including myelodysplastic syndrome (MDS), CLL, acute myeloid leukemia (AML), and NHL in clinical phase I. Luxeptinib is a non-covalent, reversible, and orally active inhibitor that targets both fms-like tyrosine kinase (FLT3) and BTK [52]. Luxeptinib effectively blocks the phosphorylation of AKT, ERK1/2, Plcg2, BTK, and S6 ribosomal proteins and significantly suppresses SYK phosphorylation in primary CLL cells [53]. This implies that Luxeptinib may be applicable to patients with different types of B-cell malignancies who have developed resistance, refractory response, or intolerance to covalent or non-covalent BTK inhibitors.
One notable method of Luxeptinib is depicted in Scheme 10 [54,55,56]. First, 7-bromo-3-oxoisoindoline-4-carbonitrile (LUXE-001) is reduced using Raney Ni to give the aldehyde LUXE-002. In ammonia, LUXE-002 is condensed with 2-oxopropanal (LUXE-003) to give the imidazole LUXE-004. The Suzuki coupling of LUXE-004 with the borate ester LUXE-005 catalyzed by Pd(PPh3)4 yields LUXE-006. Finally, 2,4,6-trifluorobenzoyl azide (LUXE-007) undergoes a Curtius rearrangement to give the isocyanate, which reacts with the amine LUXE-006 to give the urea Luxeptinib.
3.3.2. Branebrutinib (BMS-986195)
Branebrutinib, created by Bristol Myers Squibb, is currently in phase II development. This drug shows promise in treating various conditions, including atopic dermatitis (AD), Sjogren syndrome (SS), RA, and SLE. In preclinical studies, Branebrutinib has demonstrated potent inhibition of BTK activity (IC50 = 0.1 nM), leading to the suppression of B-cell activation and a subsequent reduction in disease severity [57]. The results indicate that Branebrutinib shows promise for treating autoimmune disorders such as RA and SLE [58]. Promising outcomes have been observed in clinical trials assessing the effectiveness of Branebrutinib. In a phase II trial involving individuals with relapsed or refractory MCL, Branebrutinib exhibited a high overall response rate (ORR) and long-lasting responses [59]. Additionally, in a phase III trial for RA, Branebrutinib showed significant improvement in disease activity compared to placebo.
The synthesis of Branebrutinib begins with a Pd2(dba)3-catalyzed Buchwald–Hartwig cross-coupling reaction of indole BRAN-001 with tert-butyl (S)-piperidin-3-ylcarbamate (BRAN-002) to give BRAN-003 (Scheme 11) [57]. Subsequently, BRAN-003 undergoes the hydrolysis of cyanide in the presence of H2SO4 to give the amide BRAN-004. Finally, in the presence of the condenser HATU, BRAN-004 undergoes amidation with 2-butynoic acid (TIRA-011) to obtain Branebrutinib.
3.3.3. Elsubrutinib (ABBV-105)
Elsubrutinib, developed by Abbvie Ltd., is in clinical phase II and is used to treat SLE and RA. Elsubrutinib is a BTK inhibitor that is taken orally and exhibits selectivity and irreversibility (IC50 = 0.18 μM) [60]. Elsubrutinib irreversibly inhibits BTK and exhibits superior selectivity in the kinome. It demonstrates potent activity in cellular assays involving B-cell receptors, Fc receptors, and TLR-9. Its mode of action involves the irreversible inhibition of BTK, which disrupts BCR signaling. Both preclinical and clinical research have confirmed its effectiveness and tolerable toxicity profile, positioning it as a promising treatment option for individuals with B-cell malignancies and autoimmune diseases [61]. Elsubrutinib inhibits antibody responses to both non-dependent thymus and dependent thymus antigens, reducing paw swelling and bone destruction in collagen-induced arthritis in rats. It also alleviates disease in an IFNα-accelerated lupus nephritis model [60].
The synthesis of Elsubrutinib starts with the Suzuki coupling of 4-bromo-1H-indole-7-carboxamide (ELSU-001) with the borate ester ELSU-002 catalyzed by Pd(dppf)Cl2 to obtain ELSU-003 (Scheme 12) [62]. ELSU-004 is obtained by catalytic hydrogenation of ELSU-003 with H2 and Pd/C. Subsequently, the Boc group is removed using acetyl chloride to obtain ELSU-005, which is amidated with acryloyl chloride in the presence of DIPEA to obtain ELSU-006. Finally, ELSU-006 is separated into the desired isomer of Elsubrutinib by HPLC.
3.4. Pyrazine-Fused Bicyclic Heterocycles
3.4.1. Acalabrutinib (Calquence, ACP-196)
On 31 October 2017, the FDA authorized the initial approval of Acalabrutinib to treat MCL. Subsequently, it obtained FDA approval for the treatment of CLL as well on 3 August 2022. Calquence, originally developed by AstraZeneca and Acerta Pharma LLC., is the brand name for Acalabrutinib. Acalabrutinib is a second-generation BTK inhibitor that is taken orally. It is irreversible and highly selective [63]. Acalabrutinib effectively inhibits BTK with a potency of 30 nM (IC50) and effectively suppresses CD69 B-cell activation in human whole blood with an EC50 of 8 nM. It works by binding to a Cys481 residue in the active site of BTK, forming a covalent bond, and inhibiting BTK activity [64]. Because Acalabrutinib can effectively block the activation of CD86 and CD69 downstream signaling proteins through BTK, it effectively suppresses the growth and survival of malignant B-cells. Acalabrutinib demonstrates improved selectivity and inhibition of BTK activity compared to Ibrutinib. Additionally, it exhibits significantly higher IC50 values or minimal inhibition of other kinase activities, including EGFR, ERBB4, tyrosine-protein kinase (ITK), Janus kinase 3 (JAK3), hematopoietic cell kinase (HCK), B lymphoid tyrosine kinase (BLK), feline Gardner-Rasheed sarcoma viral oncogene homolog (FGR), and SRC [65].
A method of synthesis for Acalabrutinib is summarized in Scheme 13 [66]. First, 3-chloropyrazine-2-carbonitrile (ACAL-001) is catalytically reduced by hydrogen to give the amine ACAL-002. In the presence of the condenser HATU, ACAL-002 is amidated with ((benzyloxy)carbonyl)-L-proline (ACAL-003) to give the amide ACAL-004. The cyclization of ACAL-004 in acetonitrile using phosphorus oxychloride with 1,3-dimethyl-2-imidazolidinone affords the imidazole ACAL-005. Subsequently, ACAL-005 is brominated using N-bromosuccinimide (NBS) to obtain ACAL-006. ACAL-006 undergoes a halogen aminolysis reaction in ammonia to give ACAL-007. Under the catalysis of Pd(dppf)Cl2, ACAL-007 undergoes a Suzuki coupling reaction with (4-(pyridin-2-ylcarbamoyl)phenyl)boronic acid (ACAL-008) to give ACAL-009. Treatment of ACAL-009 with 33% HBr to remove the benzyloxycarbonyl (Cbz) group gives ACAL-010. Finally, ACAL-010 is amidated with 2-butynoic acid (TIRA-011) in the presence of the condenser HATU to obtain Acalabrutinib.
3.4.2. Fenebrutinib (GDC-0853)
Fenebrutinib, a small-molecule drug, was developed by Genentech. At present, the highest research and development stage of the drug is clinical phase III, which is used to treat MS and chronic progressive MS. Fenebrutinib is a highly effective and specific non-covalent BTK inhibitor that can be taken orally. The inhibitor has Kis values of 0.91 nM, 1.6 nM, 1.3 nM, 12.6 nM, and 3.4 nM for wild-type BTK, as well as the C481S, C481R, T474I, and T474M mutants [67]. It has also shown synergy with other targeted therapies, such as Venetoclax, further enhancing its anti-tumor effects. In clinical trials, Fenebrutinib has shown promising efficacy in patients with CLL and other B-cell malignancies [68,69]. Fenebrutinib has exhibited impressive response rates and long-lasting remissions, both when used alone and in conjunction with other medications. Moreover, it has displayed promising results in patients with relapsed or refractory conditions who have not responded to prior treatments [70]. While Fenebrutinib had an acceptable safety profile, the primary end point, the SRI-4 response, was not met despite the evidence of strong pathway inhibition [68].
The preparation of Fenebrutinib is depicted in Scheme 14 [71]. First, 5-bromo-2-nitropyridine (FENE-001) undergoes Buchwald–Hartwig cross-coupling with (S)-tert-butyl 3-methylpiperazine-1-carboxylate (FENE-002) catalyzed by Pd2(dba)3 to give FENE-003. The amine FENE-004 is obtained by reducing FENE-003. A Buchwald–Hartwig cross-coupling reaction of FENE-004 with pyridinone FENE-005 catalyzed by Pd2(dba)3 affords FENE-006. Treatment of FENE-006 with HCl to remove the Boc moiety yields FENE-007. A reductive amination of FENE-007 with oxetan-3-one (FENE-008) under the reducing action of sodium cyanoborohydride yields FENE-009. Subsequently, FENE-009 undergoes a Miyaura Borylation reaction with bis(pinacolato)diboron catalyzed by Pd2(dba)3 to obtain the borate ester FENE-010. Then, FENE-010 undergoes a Suzuki coupling reaction with the intermediate FENE-011 to obtain FENE-012. Finally, the aldehyde group of FENE-012 is reduced using sodium borohydride to obtain Fenebrutinib.
3.5. Others
3.5.1. Orelabrutinib (ICP-022)
Orelabrutinib, created by Beijing InnoCare Pharma Tech, received approval from the National Medical Products Administration (NMPA) on 25 December 2020, for its effectiveness in treating MCL and CLL [72]. The NMPA also approved the drug to treat MZBL on 20 April 2023. Orelabrutinib, a potent oral inhibitor of BTK, exhibits remarkable efficacy as an antineoplastic agent. By blocking the activation of the B-cell antigen receptor signaling pathway and subsequent survival pathways triggered by BTK, it effectively inhibits the growth of cancerous B-cells with elevated levels of BTK [73]. In terms of safety, the AEs observed during the study of Orelabrutinib treatment are generally consistent with the characteristics of BTK inhibitors. The most common AEs are hematologic toxicities, including thrombocytopenia, neutropenia, and anemia. In addition, a small number of patients have also reported respiratory system infections and purpura [74].
The preparation of Orelabrutinib begins with the hydrolysis of 2,6-dichloronicotinonitrile (OREL-001) in the presence of concentrated sulfuric acid to give the amide OREL-002 (Scheme 15) [75]. Subsequently, a Suzuki coupling reaction of OREL-002 with (4-phenoxyphenyl)boronic acid (OREL-003) is catalyzed by Pd(dppf)Cl2 to obtain OREL-004, which undergoes further Suzuki coupling reaction with N-Boc-protected boronic ester OREL-005 to obtain OREL-006. Catalytic hydrogenation of OREL-006 by Pd/C gives OREL-007, which is treated with TFA to remove the Boc group. Finally, OREL-008 is N-alkylated with acryloyl chloride in the presence of TEA to obtain Orelabrutinib.
3.5.2. Pirtobrutinib (Jaypirca, LOXO-305)
Pirtobrutinib, developed by Redx Pharma, received FDA approval on 27 January 2023 to treat MCL. It is a highly selective and non-covalent-binding BTK inhibitor and demonstrates significant efficacy in suppressing different BTK-C481 substitution mutations. Pirtobrutinib exhibits a high level of selectivity for BTK, with a selectivity ratio of over 300-fold compared to the 370 other kinases tested. Additionally, at a concentration of 1 μM, it does not significantly impede any non-kinase off-targets [76,77]. Cys481 mutations play a significant role in conferring resistance to covalent BTK inhibitors, but they do not affect the efficacy of Pirtobrutinib. Although the precise mechanisms behind resistance to covalent BTK inhibitors are not yet fully understood, it is evident that these mutations play a major contributing factor [77,78,79].
The synthesis of Pirtobrutinib starts with the reaction of 5-fluoro-2-methoxybenzoic acid (PIRT-001) with thionyl chloride to obtain the chloride intermediate, and then the amidation of the chloride intermediate with 4-(aminomethyl)benzoic acid (PIRT-002) under the action of TEA to obtain amide PIRT-003 takes place (Scheme 16) [80]. Under the same conditions, PIRT-003 is reacted with sulfoxide to obtain the chloride intermediate, and then PIRT-004 is obtained by Knoevenagel condensation of the chloride intermediate with malony-dinitril. Alkylation of PIRT-004 with trimethyl orthoformate affords the ether PIRT-005. Subsequently, hydrazine PIRT-006 is transformed into the free base in TEA, followed by cyclization with PIRT-005 in the presence of TEA to give pyrazole PIRT-007. Finally, PIRT-007 is hydrolyzed in methanesulfonic acid (MsOH) to give Pirtobrutinib.
3.5.3. Tolebrutinib (SAR-442168)
Tolebrutinib, created by Principia Biopharma Inc., is currently in the advanced clinical phase III of development. This drug is specifically designed to address MS and chronic progressive MS. Tolebrutinib is a highly effective and specific inhibitor of BTK, which can be taken orally and easily crosses the BBB. In Ramos B-cells, it has shown IC50 values of 0.4 nM, while in HMC microglia cells, it has exhibited IC50 values of 0.7 nM [81]. The mechanism of action of Tolebrutinib involves blocking the activation of BTK, thereby preventing the proliferation and survival of malignant B-cells. This inhibition also leads to the suppression of downstream signaling pathways, like NF-κB and AKT, which are crucial for the growth and survival of cancer cells [82]. In clinical trials, Tolebrutinib has shown favorable efficacy and safety profiles [83]. It has demonstrated significant clinical responses in patients with relapsed or refractory CLL and MCL, leading to its accelerated approval by the FDA for these indications [84]. The drug has also shown potential in other B-cell malignancies, such as WM and DLBCL [85].
The synthetic route of Tolebrutinib as described in the publication is shown in Scheme 17 [86]. First, 2,4-dichloro-3-nitropyridine (TOLE-001) is reacted with tert-butyl (R)-3-aminopiperidine-1-carboxylate (TOLE-002) in the presence of TEA to give TOLE-003. Subsequently, TOLE-003 is reacted with bis(4-methoxybenzyl)amine (TOLE-004) in the presence of TEA to give TOLE-005, which is reduced by iron powder to give aminopyridine TOLE-006. TOLE-006 is cyclized with CDI to give TOLE-007, and treatment of TOLE-007 with TFA to remove the Boc and p-methoxybenzyl (PMB) groups gives TOLE-008. The reaction of TOLE-008 with (Boc)2O utilizes the Boc group to protect the piperidine ring to obtain TOLE-009, and the subsequent condensation of TOLE-009 with DMF-DMA to obtain TOLE-010 is performed. A Chan–Lam coupling reaction of TOLE-010 with (4-phenoxyphenyl)boronic acid (TOLE-011) catalyzed by copper acetate yields TOLE-012. TOLE-012 is stripped of its Boc group using hydrochloric acid to give TOLE-013. Finally, TOLE-013 is amidated with acryloyl chloride to obtain Tolebrutinib.
4. Challenge and Prospective
At present, the difficulties in the advancement of BTK inhibitors mainly include: (1) off-target effects and related adverse reactions and (2) drug resistance. BTK belongs to the Tec family of non-receptor tyrosine kinases and is essential for BCR signaling. Nonetheless, BTK is found in different cell types, such as macrophages and mast cells, which may lead to unintended consequences when employing small-molecule inhibitors. Achieving selectivity for BTK while avoiding inhibition of other kinases is a significant challenge in the development of these inhibitors. After iteration and optimization, the current BTK inhibitors have improved significantly in terms of off-target adverse reactions in general, but another difficulty is the problem of drug resistance, which has not been addressed in the existing commercially available BTK inhibitors. Among them, acquired resistance caused by point mutations of BTK kinase is a major cause of resistance to BTK inhibitors; moreover, covalent binding site mutations (C481S, C481F/y, and C481R), “gated region” mutations (T474I and T474S), and β-fold VII mutations (L528W) were the major ones.
In response to these problems, the pharmacochemical strategies of the new generation of BTK inhibitors focus on optimizing existing molecules, such as developing non-covalent inhibitors, avoiding steric hindrance of mutant residues, interacting with mutant residues, modifying solvent-accessible regions, and developing new skeletons. In addition, strategies to combat resistance to BTK inhibitors include combination with other targeted drugs, reduction of BTK content, inhibition of upstream and downstream pathways of BTK, combination with CAR-T cell immunotherapy, and implementation of other pathways to inhibit the proliferation of tumor cells.
5. Conclusions
In conclusion, the use of small-molecule inhibitors targeting BTK has demonstrated significant potential for treating different types of B-cell malignancies and autoimmune disorders. These inhibitors effectively hinder the function of BTK, a key player in BCR signaling and immune response. Clinical trials have demonstrated their efficacy in improving patient outcomes, including ORR and progression-free survival (PFS). Furthermore, these inhibitors have shown favorable safety profiles with manageable adverse effects. The emergence of small-molecule BTK inhibitors signifies notable progress in targeted therapy and holds immense promise for the prospective management of BTK-related diseases.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28248037/s1, Figure S1: Chemical structures of representative BTK inhibitors in the clinic; Table S1: Representative BTK inhibitors in various clinical stages. The chemical structures and clinical application of representative BTK inhibitors in diverse clinical phases are available in the supporting information (Figure S1 and Table S1).
Author Contributions
Conceptualization, Y.W. and L.Z.; resources, Q.Z.; data curation, C.W.; writing—original draft preparation, Q.Z. and C.W.; writing—review and editing, Y.W. and L.Z.; supervision, Y.W. and L.Z.; project administration, L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All of the relevant data are presented within the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Garg, N.; Padron, E.J.; Rammohan, K.W.; Goodman, C.F. Bruton’s tyrosine kinase inhibitors: The next frontier of B-cell-targeted therapies for cancer, autoimmune disorders, and multiple sclerosis. J. Clin. Med. 2022, 11, 6139. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef]
- Feng, Y.; Duan, W.; Cu, X.; Liang, C.; Xin, M. Bruton’s tyrosine kinase (BTK) inhibitors in treating cancer: A patent review (2010–2018). Expert Opin. Ther. Pat. 2019, 29, 217–241. [Google Scholar] [CrossRef]
- Zhang, D.; Gong, H.; Meng, F. Recent advances in BTK inhibitors for the treatment of inflammatory and autoimmune diseases. Molecules 2021, 26, 4907. [Google Scholar] [CrossRef]
- Alu, A.; Lei, H.; Han, X.; Wei, Y.; Wei, X. BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: Mechanisms and clinical studies. J. Hematol. Oncol. 2022, 15, 138. [Google Scholar] [CrossRef]
- de Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.L.; Chiu, H.J.; Saber, H.; Brower, M.E.; Chang, C.J.; Pfuma, E.; Habtemariam, B.; et al. FDA approval: Ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef]
- Paydas, S. Management of adverse effects/toxicity of ibrutinib. Crit. Rev. Oncol. Hematol. 2019, 136, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-generation inhibitors of Bruton tyrosine kinase. J. Hematol. Oncol. 2016, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Perutelli, F.; Montalbano, M.C.; Boccellato, E.; Coscia, M.; Vitale, C. Beyond ibrutinib: Novel BTK inhibitors for the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2022, 34, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.N.; Jimeno, A. Zanubrutinib: A new BTK inhibitor for treatment of relapsed/refractory mantle cell lymphoma. Drugs Today 2020, 56, 531–539. [Google Scholar] [CrossRef]
- Zain, R.; Vihinen, M. Structure-function relationships of covalent and non-covalent btk inhibitors. Front. Immunol. 2021, 12, 694853. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Shah, N.N.; Alencar, A.J.; Gerson, J.N.; Patel, M.R.; Fakhri, B.; Jurczak, W.; Tan, X.N.; Lewis, K.L.; Fenske, T.; et al. MCL-133 Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in previously treated mantle cell lymphoma: Updated results from the phase 1/2 BRUIN study. Clin. Lymphoma Myeloma Leuk. 2022, 22, S394–S395. [Google Scholar] [CrossRef]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef]
- Evans, E.K.; Tester, R.; Aslanian, S.; Karp, R.; Sheets, M.; Labenski, M.T.; Witowski, S.R.; Lounsbury, H.; Chaturvedi, P.; Mazdiyasni, H.; et al. Inhibition of BTK with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. [Google Scholar] [CrossRef]
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: Results from a mechanistic study. Rheumatol. Ther. 2020, 7, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Rozkiewicz, D.; Hermanowicz, J.M.; Kwiatkowska, I.; Krupa, A.; Pawlak, D. Bruton’s tyrosine kinase inhibitors (BTKIs): Review of preclinical studies and evaluation of clinical trials. Molecules 2023, 28, 2400. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.; Tester, R.W.; Kluge, A.F. 2,4-Disubstituted Pyrimidines Useful as Kinase Inhibitors. U.S. Patent 8338439B2, 25 December 2012. [Google Scholar]
- Singh, J.; Petter, R.; Wayne Tester, R.; Kluge, A.F. Heteroaryl Compounds and Uses Thereof. WO2011090760A1, 28 July 2011. [Google Scholar]
- Kelsey, R. Phase II trial of evobrutinib in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 434. [Google Scholar] [CrossRef]
- Bonafoux, D.; Davis, H.M.; Frank, K.E.; Friedman, M.M.; Herold, J.M.; Hoemann, M.Z.; Huntley, R.; Osuma, A.; Sheppard, G.; Somal, G.K.; et al. Primary Carboxamides as BTK Inhibitors. CN105530932A, 27 April 2016. [Google Scholar]
- Haselmayer, P.; Camps, M.; Liu-Bujalski, L.; Nguyen, N.; Morandi, F.; Head, J.; O’Mahony, A.; Zimmerli, S.C.; Bruns, L.; Bender, A.T.; et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J. Immunol. 2019, 202, 2888–2906. [Google Scholar] [CrossRef]
- Hodous, B.L.; Liu-Bujalski, L.; Jones, R.; Bankston, D. Compositions and Methods for the Production of Pyrimidine and Pyridine Compounds with BTK Inhibitory Activity. WO 2012170976A2, 30 October 2012. [Google Scholar]
- Angst, D.; Gessier, F.; Janser, P.; Vulpetti, A.; Wälchli, R.; Beerli, C.; Littlewood-Evans, A.; Dawson, J.; Nuesslein-Hildesheim, B.; Wieczorek, G.; et al. Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2020, 63, 5102–5118. [Google Scholar] [CrossRef]
- Kaul, M.; End, P.; Cabanski, M.; Schuhler, C.; Jakab, A.; Kistowska, M.; Kinhikar, A.; Maiolica, A.; Sinn, A.; Fuhr, R.; et al. Remibrutinib (LOU064): A selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin. Transl. Sci. 2021, 14, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Berger, W.; Giménez-Arnau, A.; Hayama, K.; Jain, V.; Reich, A.; Haemmerle, S.; Lheritier, K.; Walsh, P.; Xia, S.; et al. Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria. J. Allergy Clin. Immunol. 2022, 150, 1498–1506.E2. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Brown, J.R. Ibrutinib: A first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014, 10, 957–967. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef]
- Xu, X. Method for Preparing Ibrutinib. CN103626774A, 12 March 2014. [Google Scholar]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef]
- St-Pierre, F.; Ma, S. Use of BTK inhibitors in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): A practical guidance, Blood Lymphat. Cancer 2022, 12, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.X.; Zhu, H.Y.; Li, X.T.; Xia, Y.; Miao, K.R.; Zhao, S.S.; Wu, Y.J.; Wang, L.; Xu, W.; Li, J.Y. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400. [Google Scholar] [CrossRef]
- Zhiwei, W.; Yunhang, G. Fused Heterocyclic Compounds as Protein Kinase Inhibitors. U.S. Patent 20170073349A1, 16 March 2017. [Google Scholar]
- Dhillon, S. Tirabrutinib: First approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kong, H.; Li, C.; Dong, X.; Wu, Y.; Zhuang, Y.; Han, S.; Lei, T.; Yang, H. Bruton’s tyrosine kinase inhibitors in primary central nervous system lymphoma-evaluation of anti-tumor efficacy and brain distribution. Transl. Cancer Res. 2021, 10, 1975–1983. [Google Scholar] [CrossRef]
- Walter, H.S.; Jayne, S.; Rule, S.A.; Cartron, G.; Morschhauser, F.; Macip, S.; Karlin, L.; Jones, C.; Herbaux, C.; Quittet, P.; et al. Long-term follow-up of patients with CLL treated with the selective Bruton’s tyrosine kinase inhibitor ONO/GS-4059. Blood 2017, 129, 2808–2810. [Google Scholar] [CrossRef] [PubMed]
- Munakata, W.; Ando, K.; Hatake, K.; Fukuhara, N.; Kinoshita, T.; Fukuhara, S.; Shirasugi, Y.; Yokoyama, M.; Ichikawa, S.; Ohmachi, K.; et al. Phase I study of tirabrutinib (ONO-4059/GS-4059) in patients with relapsed or refractory B-cell malignancies in Japan. Cancer Sci. 2019, 110, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yoshizawa, T. Purinone Derivative. WO2011152351A1, 2011. [Google Scholar]
- Woyach, J.; Flinn, I.W.; Awan, F.T.; Eradat, H.; Brander, D.; Tees, M.; Parikh, S.A.; Phillips, T.; Wang, W.; Reddy, N.M.; et al. P682: Nemtabrutinib (MK-1026), a non-covalent inhibitor of wild-type and C481s mutated Bruton tyrosine kinase for B-cell malignancies: Efficacy and safety of the phase 2 dose-expansion bellwave-001 study. HemaSphere 2022, 6, 578–579. [Google Scholar] [CrossRef]
- Woyach, J.A.; Flinn, I.W.; Awan, F.T.; Eradat, H.; Brander, D.; Tees, M.; Parikh, S.A.; Phillips, T.J.; Ghori, R.; Reddy, N.M.; et al. Efficacy and safety of nemtabrutinib, a wild-type and C481S-mutated Bruton tyrosine kinase inhibitor for B-Cell malignancies: Updated analysis of the open-label phase 1/2 dose-expansion bellwave-001 study. Blood 2022, 140, 7004–7006. [Google Scholar] [CrossRef]
- Muhowski, E.M.; Ravikrishnan, J.; Gordon, B.; Yu, L.; Misra, S.; Walker, B.; Eathiraj, S.; Sampath, D.; Rogers, K.A.; Byrd, J.C.; et al. Preclinical evaluation of combination nemtabrutinib and venetoclax in chronic lymphocytic leukemia. J. Hematol. Oncol. 2022, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Corry, J.; Desmond, R.; Di Maso, M.J. Synthesis of BTK Inhibitor and Intermediates Thereof. WO2022251404A1, 1 December 2022. [Google Scholar]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Hill, R.; Bradshaw, J.; Bisconte, A.; Tam, D.; Owens, T.; Brameld, K.; Smith, P.; Funk, J.; Goldstein, D.; Nunn, P. Preclinical characterization of PRN1008, a novel reversible covalent inhibitor of BTK that shows efficacy in a RAT model of collagen-induced arthritis. Ann. Rheum. Dis. 2015, 74, 216. [Google Scholar] [CrossRef]
- Owens, T.D.; Brameld, K.A.; Verner, E.J.; Ton, T.; Li, X.; Zhu, J.; Masjedizadeh, M.R.; Bradshaw, J.M.; Hill, R.J.; Tam, D.; et al. Discovery of reversible covalent Bruton’s tyrosine kinase inhibitors PRN473 and PRN1008 (rilzabrutinib). J. Med. Chem. 2022, 65, 5300–5316. [Google Scholar] [CrossRef]
- Murrell, D.F.; Patsatsi, A.; Stavropoulos, P.; Baum, S.; Zeeli, T.; Kern, J.S.; Roussaki-Schulze, A.V.; Sinclair, R.; Bassukas, I.D.; Thomas, D.; et al. Proof of concept for the clinical effects of oral rilzabrutinib, the first Bruton tyrosine kinase inhibitor for pemphigus vulgaris: The phase II BELIEVE study. Br. J. Dermatol. 2021, 185, 745–755. [Google Scholar] [CrossRef]
- Owens, T.; Verner, E. Pyrazolopyrimidine Compounds as Kinase Inhibitors. WO2014039899A1, 3 March 2014. [Google Scholar]
- Mao, L.; Tang, W.; Zhang, X.; Liu, J.; Chen, Y.; Hua, Y.; Weng, B.; Mo, X.; Bao, Y.; Teng, L.; et al. Discovery of a novel, selective and irreversible inhibitor (abivertinib) of mutated EGFR and T790M-induced resistance for the treatment of NSCLC. Med. Drug Discov. 2020, 6, 100035. [Google Scholar] [CrossRef]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (review). Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Zhao, L.; Xi, B. Novel Pyrrolopyrimidine Compounds as Inhibitors of Protein Kinases. U.S. Patent 20150210702A1, 30 July 2015. [Google Scholar]
- Rice, W.G.; Howell, S.B.; Zhang, H.; Rastgoo, N.; Local, A.; Kurtz, S.E.; Lo, P.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; et al. Luxeptinib (CG-806) targets FLT3 and clusters of kinases operative in acute myeloid leukemia. Mol. Cancer Ther. 2022, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Zhang, H.; Sivina, M.; Vaca, A.; Thompson, P.A.; Jain, N.; Ferrajoli, A.; Estrov, Z.E.; Keating, M.J.; Wierda, W.G.; et al. CG-806, a first-in-class pan-FLT3/pan-BTK Inhibitor, exhibits broad signaling inhibition in chronic lymphocytic leukemia cells. Blood 2019, 134, 3051. [Google Scholar] [CrossRef]
- Cho, J.M.; Hong, Y. Methods for Treating Patients with Hematologic Malignancies. WO2018156578A1, 30 August 2018. [Google Scholar]
- Hong, Y.R.; Na, J.E.; Min, I.S.; Cha, H.J. 2,3-Dihydro-isoindole-1-on Derivative as BTK Kinase Suppressant, and Pharmaceutical Composition Including Same. U.S. Patent 20150336934A1, 26 November 2015. [Google Scholar]
- Rice, W.G.; Cho, J.M.; Hong, Y. Methods for Treating Patients with Hematologic Malignancies. CN110621665A, 27 December 2019. [Google Scholar]
- Watterson, S.H.; Liu, Q.; Beaudoin Bertrand, M.; Batt, D.G.; Li, L.; Pattoli, M.A.; Skala, S.; Cheng, L.; Obermeier, M.T.; Moore, R.; et al. Discovery of branebrutinib (BMS-986195): A strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. [Google Scholar] [CrossRef] [PubMed]
- Catlett, I.M.; Nowak, M.; Kundu, S.; Zheng, N.; Liu, A.; He, B.; Girgis, I.G.; Grasela, D.M. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo-controlled trial in healthy participants. Br. J. Clin. Pharmacol. 2020, 86, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Goess, C.; Harris, C.M.; Murdock, S.; McCarthy, R.W.; Sampson, E.; Twomey, R.; Mathieu, S.; Mario, R.; Perham, M.; Goedken, E.R.; et al. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. 2019, 29, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Friedman, A.; Drescher, E.; Singhal, A.; Cortes-Maisonet, G.; Doan, T.; Lu, W.; Wang, Z.; Nader, A.; Housley, W.; et al. Safety and efficacy of elsubrutinib or upadacitinib alone or in combination (ABBV-599) in patients with rheumatoid arthritis and inadequate response or intolerance to biological therapies: A multicentre, double-blind, randomised, controlled, phase 2 trial. Lancet Rheumatol. 2022, 4, E395–E406. [Google Scholar] [CrossRef]
- Bonafoux, D.; Davis, H.M.; Frank, K.E.; Friedman, M.M. Primary Carboxamides as BTK Inhibitors. WO2014210255A1, 31 December 2014. [Google Scholar]
- Wu, J.; Zhang, M.; Liu, D. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 21. [Google Scholar] [CrossRef]
- Cheah, C.Y.; Seymour, J.F.; Wang, M.L. Mantle Cell Lymphoma. J. Clin. Oncol. 2016, 34, 1256–1269. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Montraveta, A.; Niemann, C.U.; Mora-Jensen, H.; Gulrajani, M.; Krantz, F.; Mantel, R.; Smith, L.L.; McClanahan, F.; Harrington, B.K.; et al. The bruton tyrosine kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin. Cancer Res. 2017, 23, 2831–2841. [Google Scholar] [CrossRef]
- Barf, T.A.; Gerardus, C.J.; Maria, J.; Man, D.A.; Antonius, P.; Oubrie, A.A. Oubrie. 4-Imidazopyridazin-1-yl-benzamides and 4-imidazotriazin-1-yl-benzamides as BTK-Inhibitors. WO2013010868A1, 24 January 2013. [Google Scholar]
- Erickson, R.I.; Schutt, L.K.; Tarrant, J.M.; McDowell, M.; Liu, L.; Johnson, A.R.; Lewin-Koh, S.C.; Hedehus, M.; Ross, J.; Carano, R.A.; et al. Bruton’s tyrosine kinase small molecule inhibitors induce a distinct pancreatic toxicity in rats. J. Pharmacol. Exp. Ther. 2017, 360, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, safety, and pharmacodynamic effects of the Bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: Results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Christopher, H.; Meire, B.; Alexandra, G.; James, C.; Adam, J.; Amit, B.-O. Fenebrutinib demonstrates the highest potency of Bruton tyrosine kinase inhibitors (BTKis) in phase 3 clinical development for multiple sclerosis (MS). Neurology 2021, 96, 4437. [Google Scholar]
- Castillo, J.J.; Treon, S.P. What is new in the treatment of Waldenstrom macroglobulinemia? Leukemia 2019, 33, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.J.; Johnson, A.R.; Misner, D.L.; Belmont, L.D.; Castanedo, G.; Choy, R.; Coraggio, M.; Dong, L.; Eigenbrot, C.; Erickson, R.; et al. Discovery of GDC-0853: A potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 2018, 61, 2227–2245. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Orelabrutinib: First approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Song, Y.; Li, Z.; Yang, S.; Liu, L.; Hu, Y.; Zhang, W.; Zhou, J.; Gao, S.; Ding, K. Safety, tolerability and efficacy of orelabrutinib, once a day, to treat Chinese patients with relapsed or refractory chronic lymphocytic leukemia/small cell leukemia. Blood 2019, 134, 4319. [Google Scholar] [CrossRef]
- Gu, D.; Li, J.; Miao, Y. Evaluating orelabrutinib as a novel treatment option for relapsed/refractory chronic lymphocytic leukemia in China. Expert Opin. Pharmacother. 2022, 23, 1979–1986. [Google Scholar] [CrossRef]
- Xiangyang, C.; Yingxiang, G.; Chong, L.; Haihong, N. Substituted Nicotinimide Inhibitors of btk and Their Preparation and Use in the Treatment of Cancer, Inflammation and Autoimmune Disease. WO2015048662A2, 2 April 2015. [Google Scholar]
- Gomez, E.B.; Rosendahal, M.S.; Rothenberg, S.M.; Andrews, S.W.; Brandhuber, B.J. Loxo-305, a highly selective and non-covalent next generation BTK inhibitor, inhibits diverse BTK C481 substitution mutations. Blood 2019, 134, 4644. [Google Scholar] [CrossRef]
- Jensen, J.L.; Mato, A.R.; Pena, C.; Roeker, L.E.; Coombs, C.C. The potential of pirtobrutinib in multiple B-cell malignancies. Ther. Adv. Hematol. 2022, 13, 20406207221101697. [Google Scholar] [CrossRef]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.D.A.; Charles, E.; Jared, F.; Scott, F.; Nicholas, M. Processes and Intermediates for the Preparation of (S)-5-amino-3-(4-((5-fluoro-2-methoxybenzamido)methyl)phenyl)-1-(1,1,1-trifluoropropane-2-yl)-1H-pyrazole-4-carboxamide. WO2022056100A1, 17 March 2022. [Google Scholar]
- Francesco, M.; Wong, M.; LaStant, J.; Finkle, D.; Loewenstein, N.; Macsata, R.; Lindstrom, M.; Shu, J.; Ton, T.; Zhu, J. PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult. Scler. J. 2017, 23, 989. [Google Scholar]
- Ringheim, G.E.; Wampole, M.; Oberoi, K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: Making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front. Immunol. 2021, 12, 662223. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, R.; Gajofatto, A. Tolebrutinib. Bruton tyrosine kinase (BTK) inhibitor treatment of multiple sclerosis. Drugs Future 2022, 47, 325–336. [Google Scholar] [CrossRef]
- Kueffer, L.E.; Joseph, R.E.; Andreotti, A.H. Reining in BTK: Interdomain interactions and their importance in the regulatory control of BTK. Front. Cell Dev. Biol. 2021, 9, 655489. [Google Scholar] [CrossRef]
- Goldstein, D.M.; Owens, T.D. Tyrosine Kinase Inhibitors. CN106459049A, 22 February 2017. [Google Scholar]
Figure 1.
Signaling pathway of BTK. p: phosphorylation.

Scheme 1.
Synthesis of Spebrutinib.

Scheme 2.
Synthesis of Evobrutinib.

Scheme 3.
Synthesis of Remibrutinib.

Scheme 4.
Synthesis of Ibrutinib.

Scheme 5.
Synthesis of Zanubrutinib.

Scheme 6.
Synthesis of Tirabrutinib Hydrochloride.

Scheme 7.
Synthesis of Nemtabrutinib.

Scheme 8.
Synthesis of Rilzabrutinib.

Scheme 9.
Synthesis of Abivertinib.

Scheme 10.
Synthesis of Luxeptinib.

Scheme 11.
Synthesis of Branebrutinib.

Scheme 12.
Synthesis of Elsubrutinib.

Scheme 13.
Synthesis of Acalabrutinib.

Scheme 14.
Synthesis of Fenebrutinib.

Scheme 15.
Synthesis of Orelabrutinib.

Scheme 16.
Synthesis of Pirtobrutinib.

Scheme 17.
Synthesis of Tolebrutinib.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Q.; Wen, C.; Zhao, L.; Wang, Y. A Comprehensive Review of Small-Molecule Inhibitors Targeting Bruton Tyrosine Kinase: Synthetic Approaches and Clinical Applications. Molecules 2023, 28, 8037. https://doi.org/10.3390/molecules28248037
AMA Style
Zhang Q, Wen C, Zhao L, Wang Y. A Comprehensive Review of Small-Molecule Inhibitors Targeting Bruton Tyrosine Kinase: Synthetic Approaches and Clinical Applications. Molecules. 2023; 28(24):8037. https://doi.org/10.3390/molecules28248037
Chicago/Turabian StyleZhang, Qi, Changming Wen, Lijie Zhao, and Yatao Wang. 2023. "A Comprehensive Review of Small-Molecule Inhibitors Targeting Bruton Tyrosine Kinase: Synthetic Approaches and Clinical Applications" Molecules 28, no. 24: 8037. https://doi.org/10.3390/molecules28248037