
Degree of Fibrosis in Human Atrial Tissue Is Not the Hallmark Driving AF

 ,
,  ,
,  , , , , and
, , , , and 
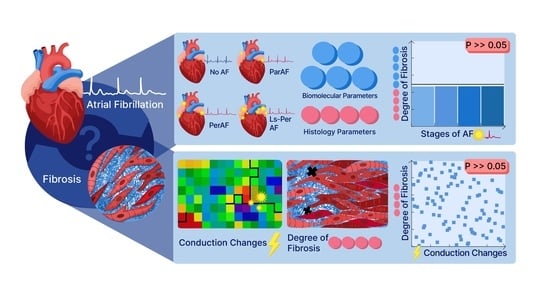
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Blood and Tissue Sampling
2.3. Mapping Procedure
2.4. Tissue Analysis for Western Blot and Immunohistochemistry
2.5. Serum Analysis for ELISA
2.6. Analysis of Mapping Data
2.7. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Total, Endomysial, and Perimysial Fibrosis Levels, and Cardiomyocyte Diameter between the Various Stages of AF
3.3. Determination of Additional Fibrosis Markers αSMA and TIMP in Various Stages of AF
3.4. Determination of Additional Fibrosis Markers NCAM and LOX in Various Stages of AF
3.5. Serum Fibrosis Marker: Ratio CITP:MMP1
3.6. Correlation between Histological Fibrosis Markers and Electrophysiology
3.7. Spatial Analysis of Electrophysiological and Histological Staining
4. Discussion
4.1. Quantification of Biomolecular Markers for Atrial Fibrosis
4.2. Electrophysiology and Atrial Fibrosis in RAA
4.3. Spatial Analyses of Atrial Fibrosis: Histological and Electrophysiological Approach
4.4. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Haissaguerre, M.; Jais, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Metayer, P.; Clementy, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998, 339, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef]
- de Groot, N.M.S.; Allessie, M.A. Pathophysiology of atrial fibrillation: Focal patterns of activation. Pacing Clin. Electrophysiol. 2019, 42, 1312–1319. [Google Scholar] [CrossRef]
- Sim, I.; Bishop, M.; O’Neill, M.; Williams, S.E. Left atrial voltage mapping: Defining and targeting the atrial fibrillation substrate. J. Interv. Card Electrophysiol. 2019, 56, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef]
- de Jong, S.; van Veen, T.A.; van Rijen, H.V.; de Bakker, J.M. Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharm. 2011, 57, 630–638. [Google Scholar] [CrossRef]
- Li, D.; Fareh, S.; Leung, T.K.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Calderone, A.; Bel-Hadj, S.; Drapeau, J.; El-Helou, V.; Gosselin, H.; Clement, R.; Villeneuve, L. Scar myofibroblasts of the infarcted rat heart express natriuretic peptides. J. Cell Physiol. 2006, 207, 165–173. [Google Scholar] [CrossRef]
- Ausma, J.; Litjens, N.; Lenders, M.H.; Duimel, H.; Mast, F.; Wouters, L.; Ramaekers, F.; Allessie, M.; Borgers, M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J. Mol. Cell Cardiol. 2001, 33, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [Green Version]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef]
- Al-U’datt, D.; Allen, B.G.; Nattel, S. Role of the lysyl oxidase enzyme family in cardiac function and disease. Cardiovasc. Res. 2019, 115, 1820–1837. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Hermida, N.; Valencia, F.; de Teresa, E.; Diez, J. Role of lysyl oxidase in myocardial fibrosis: From basic science to clinical aspects. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1–H9. [Google Scholar] [CrossRef] [Green Version]
- Nagao, K.; Sowa, N.; Inoue, K.; Tokunaga, M.; Fukuchi, K.; Uchiyama, K.; Ito, H.; Hayashi, F.; Makita, T.; Inada, T.; et al. Myocardial expression level of neural cell adhesion molecule correlates with reduced left ventricular function in human cardiomyopathy. Circ. Heart Fail. 2014, 7, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Verheule, S.; Eckstein, J.; Linz, D.; Maesen, B.; Bidar, E.; Gharaviri, A.; Schotten, U. Role of endo-epicardial dissociation of electrical activity and transmural conduction in the development of persistent atrial fibrillation. Prog. Biophys. Mol. Biol. 2014, 115, 173–185. [Google Scholar] [CrossRef]
- Robinson, T.F.; Factor, S.M.; Sonnenblick, E.H. The heart as a suction pump. Sci. Am. 1986, 254, 84–91. [Google Scholar] [CrossRef]
- Sohns, C.; Marrouche, N.F. Atrial fibrillation and cardiac fibrosis. Eur. Heart J. 2020, 41, 1123–1131. [Google Scholar] [CrossRef]
- van Brakel, T.J.; van der Krieken, T.; Westra, S.W.; van der Laak, J.A.; Smeets, J.L.; van Swieten, H.A. Fibrosis and electrophysiological characteristics of the atrial appendage in patients with atrial fibrillation and structural heart disease. J. Interv. Card Electrophysiol. 2013, 38, 85–93. [Google Scholar] [CrossRef]
- Kottkamp, H. Human atrial fibrillation substrate: Towards a specific fibrotic atrial cardiomyopathy. Eur. Heart J. 2013, 34, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Lanters, E.A.; van Marion, D.M.; Kik, C.; Steen, H.; Bogers, A.J.; Allessie, M.A.; Brundel, B.J.; de Groot, N.M. HALT & REVERSE: Hsf1 activators lower cardiomyocyt damage; towards a novel approach to REVERSE atrial fibrillation. J. Transl. Med. 2015, 13, 347. [Google Scholar] [CrossRef] [Green Version]
- Yaksh, A.; van der Does, L.J.; Kik, C.; Knops, P.; Oei, F.B.; van de Woestijne, P.C.; Bekkers, J.A.; Bogers, A.J.; Allessie, M.A.; de Groot, N.M. A novel intra-operative, high-resolution atrial mapping approach. J. Interv. Card Electrophysiol. 2015, 44, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Teuwen, C.P.; Yaksh, A.; Lanters, E.A.; Kik, C.; van der Does, L.J.; Knops, P.; Taverne, Y.J.; van de Woestijne, P.C.; Oei, F.B.; Bekkers, J.A.; et al. Relevance of Conduction Disorders in Bachmann’s Bundle during Sinus Rhythm in Humans. Circ. Arrhythm. Electrophysiol. 2016, 9, e003972. [Google Scholar] [CrossRef] [Green Version]
- Mouws, E.; Lanters, E.A.H.; Teuwen, C.P.; van der Does, L.; Kik, C.; Knops, P.; Bekkers, J.A.; Bogers, A.; de Groot, N.M.S. Epicardial Breakthrough Waves during Sinus Rhythm: Depiction of the Arrhythmogenic Substrate? Circ. Arrhythm Electrophysiol. 2017, 10, e005145. [Google Scholar] [CrossRef]
- Kik, C.; Mouws, E.; Bogers, A.; de Groot, N.M.S. Intra-operative mapping of the atria: The first step towards individualization of atrial fibrillation therapy? Expert Rev. Cardiovasc. 2017, 15, 537–545. [Google Scholar] [CrossRef]
- Lanters, E.A.H.; Yaksh, A.; Teuwen, C.P.; van der Does, L.; Kik, C.; Knops, P.; van Marion, D.M.S.; Brundel, B.; Bogers, A.; Allessie, M.A.; et al. Spatial distribution of conduction disorders during sinus rhythm. Int. J. Cardiol. 2017, 249, 220–225. [Google Scholar] [CrossRef]
- Heida, A.; van der Does, W.F.B.; van Staveren, L.N.; Taverne, Y.; Roos-Serote, M.C.; Bogers, A.; de Groot, N.M.S. Conduction Heterogeneity: Impact of Underlying Heart Disease and Atrial Fibrillation. JACC Clin. Electrophysiol. 2020, 6, 1844–1854. [Google Scholar] [CrossRef]
- Schram-Serban, C.; Heida, A.; Roos-Serote, M.C.; Knops, P.; Kik, C.; Brundel, B.; Bogers, A.; de Groot, N.M.S. Heterogeneity in Conduction Underlies Obesity-Related Atrial Fibrillation Vulnerability. Circ. Arrhythm Electrophysiol. 2020, 13, e008161. [Google Scholar] [CrossRef]
- van Schie, M.S.; Kharbanda, R.K.; Houck, C.A.; Lanters, E.A.H.; Taverne, Y.; Bogers, A.; de Groot, N.M.S. Identification of Low-Voltage Areas: A Unipolar, Bipolar, and Omnipolar Perspective. Circ. Arrhythm Electrophysiol. 2021, 14, e009912. [Google Scholar] [CrossRef]
- van Schie, M.S.; Heida, A.; Taverne, Y.; Bogers, A.; de Groot, N.M.S. Identification of local atrial conduction heterogeneities using high-density conduction velocity estimation. Europace 2021. [Google Scholar] [CrossRef]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Ausma, J.; Wijffels, M.; Thone, F.; Wouters, L.; Allessie, M.; Borgers, M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 1997, 96, 3157–3163. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Bellocci, F.; Morgante, E.; Russo, M.A.; Maseri, A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997, 96, 1180–1184. [Google Scholar] [CrossRef]
- Platonov, P.G.; Mitrofanova, L.B.; Orshanskaya, V.; Ho, S.Y. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J. Am. Coll Cardiol. 2011, 58, 2225–2232. [Google Scholar] [CrossRef] [Green Version]
- van Schie, M.S.; Starreveld, R.; Roos-Serote, M.C.; Taverne, Y.; van Schaagen, F.R.N.; Bogers, A.; de Groot, N.M.S. Classification of sinus rhythm single potential morphology in patients with mitral valve disease. Europace 2020, 22, 1509–1519. [Google Scholar] [CrossRef]
- Aguiar, C.M.; Gawdat, K.; Legere, S.; Marshall, J.; Hassan, A.; Kienesberger, P.C.; Pulinilkunnil, T.; Castonguay, M.; Brunt, K.R.; Legare, J.F. Fibrosis independent atrial fibrillation in older patients is driven by substrate leukocyte infiltration: Diagnostic and prognostic implications to patients undergoing cardiac surgery. J. Transl. Med. 2019, 17, 413. [Google Scholar] [CrossRef]
- Spronk, H.M.; De Jong, A.M.; Verheule, S.; De Boer, H.C.; Maass, A.H.; Lau, D.H.; Rienstra, M.; van Hunnik, A.; Kuiper, M.; Lumeij, S.; et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar] [CrossRef]
- Verheule, S.; Tuyls, E.; Gharaviri, A.; Hulsmans, S.; van Hunnik, A.; Kuiper, M.; Serroyen, J.; Zeemering, S.; Kuijpers, N.H.; Schotten, U. Loss of continuity in the thin epicardial layer because of endomysial fibrosis increases the complexity of atrial fibrillatory conduction. Circ. Arrhythm Electrophysiol. 2013, 6, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Shiroshita-Takeshita, A.; Brundel, B.J.; Burstein, B.; Leung, T.K.; Mitamura, H.; Ogawa, S.; Nattel, S. Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc Res. 2007, 74, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 2004, 109, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattenlohner, S.; Waller, C.; Ertl, G.; Bultmann, B.D.; Muller-Hermelink, H.K.; Marx, A. NCAM(CD56) and RUNX1(AML1) are up-regulated in human ischemic cardiomyopathy and a rat model of chronic cardiac ischemia. Am. J. Pathol. 2003, 163, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Adam, O.; Theobald, K.; Lavall, D.; Grube, M.; Kroemer, H.K.; Ameling, S.; Schafers, H.J.; Bohm, M.; Laufs, U. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J. Mol. Cell Cardiol. 2011, 50, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Song, Y.; Song, Y.; Yuan, J.; Cui, J.; Zhao, S.; Qiao, S. Collagen Cross-Linking Is Associated With Cardiac Remodeling in Hypertrophic Obstructive Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e017752. [Google Scholar] [CrossRef] [PubMed]
- Ravassa, S.; Lopez, B.; Querejeta, R.; Echegaray, K.; San Jose, G.; Moreno, M.U.; Beaumont, F.J.; Gonzalez, A.; Diez, J. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J. Hypertens. 2017, 35, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kleber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.L.; Jensen, H.K.; Peel, S.A.; Chiribiri, A.; Grondal, A.K.; Bloch, L.O.; Pedersen, S.F.; Bentzon, J.F.; Kolbitsch, C.; Karim, R.; et al. Cardiac magnetic resonance and electroanatomical mapping of acute and chronic atrial ablation injury: A histological validation study. Eur. Heart J. 2014, 35, 1486–1495. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | ParAF | PerAF | LSPerAF | AF Total | p * | p † | |
|---|---|---|---|---|---|---|---|
| Number of patients | 48 | 22 | 28 | 17 | 67 | ||
| Male | 34 (70.8) | 15 (68.2) | 19 (67.9) | 15 (88.2) | 49 (73.1) | ||
| Age (years) | 67.20 ± 12.12 | 68.28 ± 12.95 | 66.58 ± 8.54 | 71.80 ± 5.85 | 68.47 | ||
| BMI | 27.92 (20.42–38.2) | 25.23 (18.81–35.66) | 26.47 (19.25–37.56) | 28.37 (23.84–34.72) | 26.45 (18.81–37.56) | ||
| Hypertension | 31 (64.6) | 17 (77.3) | 16 (57.1) | 10 (58.8) | 43 (64.2) | ||
| Dyslipidemia | 19 (39.6) | 11 (50) | 6 (21.4) | 3 (17.6) | 20 (29.9) | ||
| DM | 15 (31.3) | 2 (9.1) | 4 (14.3) | 6 (35.3) | 12 (17.9) | ||
| Thyroid disease | 4 (8.3) | 3 (13.6) | 2 (7.1) | 1 (5.9) | 6 (9) | ||
| CHD | 2 (4.2) | 3 (13.6) | 5 (17.9) | 1 (5.9) | 9 (13.4) | ||
| AVD | 4 (8.3) | 4 (18.2) | 5 (17.9) | 3 (17.6) | 12 (17.9) | 0.0221 | |
| CABG | 29 (60.4) | 4 (18.2) | 2 (7.1) | 4 (23.5) | 10 (14.9) | ||
| MVD | 2 (4.2) | 3 (13.6) | 12 (42.9) | 5 (9.4) | 20 (29.9) | 0.0156 | |
| MAZE | 0 | 2 (9.1) | 0 | 0 | 2 (3) | ||
| AVD + CABG | 9 (18.8) | 4 (18.2) | 2 (7.1) | 2 (11.8) | 8 (11.9) | ||
| MVD + CABG-TVR | 2 (4.2) 0 | 2 (9.1) 0 | 2 (7.1) 0 | 1 (5.9) 1 (5.9) | 5 (7.5) 1 (1.5) | ||
| LV Function | |||||||
| -normal | 39 (81.3) | 19 (86.4) | 16 (57.1) | 10 (58.8) | 45 (67.2) | ||
| -mild imp. | 9 (18.8) | 2 (9.1) | 6 (21.4) | 5 (29.4) | 13 (19.4) | ||
| -moderate imp. | 0 | 1 (4.5) | 5 (17.9) | 2 (11.8) | 8 (11.9) | ||
| -severe imp. | 0 | 0 | 1 (3.6) | 0 | 1 (1.5) | ||
| Use of anti-arrhythmic drug | |||||||
| -Class I | 1 (2.1) | 2 (9.1) | 1 (3.6) | 0 | 3 (4.5) | ||
| -Class II | 33 (68.8) | 12 (54.5) | 20 (71.4) | 14 (82.4) | 46 (68.7) | ||
| -Class III | 0 | 7 (31.8) | 5 (17.9) | 1 (5.9) | 13 (19.4) | 7 × 10−4 | 3.3×10−3 |
| -Class IV | 3 (6.3) | 0 | 1 (3.6) | 2 (11.8) | 3 (4.5) | ||
| Digoxin | 0 | 1 (4.5) | 9 (32.1) | 5 (29.4) | 15 (22.4) | 7.8 × 10−5 | 1.2 × 10−3 |
| Use of statin | 35 (72.9) | 13 (59.1) | 9 (32.1) | 12 (70.6) | 34 (50.7) | ||
| Use ACE, ARB, AT2 antagonist | 30 (62.5) | 12 (54.5) | 18 (64.3) | 14 (82.4) | 44 (65.7) | 4.2 × 10−3 | 2.8 × 10−2 |
| -missing value | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Ramos, K.; Pool, L.; van Schie, M.S.; Wijdeveld, L.F.J.M.; van der Does, W.F.B.; Baks, L.; Sultan, H.M.D.; van Wijk, S.W.; Bogers, A.J.J.C.; Verheule, S.; et al. Degree of Fibrosis in Human Atrial Tissue Is Not the Hallmark Driving AF. Cells 2022, 11, 427. https://doi.org/10.3390/cells11030427
S. Ramos K, Pool L, van Schie MS, Wijdeveld LFJM, van der Does WFB, Baks L, Sultan HMD, van Wijk SW, Bogers AJJC, Verheule S, et al. Degree of Fibrosis in Human Atrial Tissue Is Not the Hallmark Driving AF. Cells. 2022; 11(3):427. https://doi.org/10.3390/cells11030427
Chicago/Turabian StyleS. Ramos, Kennedy, Lisa Pool, Mathijs S. van Schie, Leonoor F. J. M. Wijdeveld, Willemijn F. B. van der Does, Luciënne Baks, H. M. Danish Sultan, Stan W. van Wijk, Ad J. J. C. Bogers, Sander Verheule, and et al. 2022. "Degree of Fibrosis in Human Atrial Tissue Is Not the Hallmark Driving AF" Cells 11, no. 3: 427. https://doi.org/10.3390/cells11030427