Author Contributions
Conceptualization, M.A.L.-G.; Methodology, B.Y.C.; Software, B.Y.C.; Validation, J.L.P., L.A.G.-P., Y.J.A. and J.S.C.; Formal analysis, J.L.P., Y.J.A. and M.A.L.-G.; Investigation, B.Y.C. and M.A.L.-G.; Data curation, J.L.P. and L.A.G.-P.; Writing—original draft, M.A.L.-G.; Writing—review & editing, J.L.P., L.A.G.-P., Y.J.A. and J.S.C.; Visualization, M.A.L.-G.; Supervision, J.L.P.; Funding acquisition, J.S.C. All authors have read and agreed to the published version of the manuscript.
Scheme 1.
Skeleton of the flavylium ion of an anthocyanin.
Scheme 1.
Skeleton of the flavylium ion of an anthocyanin.
Figure 1.
Structure of the model after applying a theory level M062X/3-21g* (gd3bj).
Figure 1.
Structure of the model after applying a theory level M062X/3-21g* (gd3bj).
Figure 2.
Initial random structures generated by the Molclus program.
Figure 2.
Initial random structures generated by the Molclus program.
Figure 3.
Non-covalent interaction isosurfaces calculated using VDM and MULTIWFN softwares. The colored graph next to it represents all interactions in 2D. Blue spikes (attractions), green spikes (non-covalent), and red spikes (repulsions). (A) Calculations using M06-2X/gd3bj. (B) Calculations using ωB97X-D and (C) calculations using APFD. All under the same basis set 6-31g+(d,p).
Figure 3.
Non-covalent interaction isosurfaces calculated using VDM and MULTIWFN softwares. The colored graph next to it represents all interactions in 2D. Blue spikes (attractions), green spikes (non-covalent), and red spikes (repulsions). (A) Calculations using M06-2X/gd3bj. (B) Calculations using ωB97X-D and (C) calculations using APFD. All under the same basis set 6-31g+(d,p).
Figure 4.
Cyanidin: UV spectrum with the three strongest transitions calculated at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 4.
Cyanidin: UV spectrum with the three strongest transitions calculated at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 5.
Calculated UV transitions of cyanidin at the ωB97X-D/6-31g+(d,p) theory level.
Figure 5.
Calculated UV transitions of cyanidin at the ωB97X-D/6-31g+(d,p) theory level.
Figure 6.
Calculated UV transitions of resveratrol at ωB97X-D/6-31g+(d,p) theory level.
Figure 6.
Calculated UV transitions of resveratrol at ωB97X-D/6-31g+(d,p) theory level.
Figure 7.
Resveratrol: UV spectrum with the three strongest transitions calculated. λmax 343.56 nm at ωB97X-D/6-31g+(d,p) level of theory.
Figure 7.
Resveratrol: UV spectrum with the three strongest transitions calculated. λmax 343.56 nm at ωB97X-D/6-31g+(d,p) level of theory.
Figure 8.
Complex: resveratrol–cyanidin. UV spectrum with the three strongest transitions calculated. λmax 468.83 nm at ωB97X-D/6-31g+(d,p) level of theory.
Figure 8.
Complex: resveratrol–cyanidin. UV spectrum with the three strongest transitions calculated. λmax 468.83 nm at ωB97X-D/6-31g+(d,p) level of theory.
Figure 9.
MO and calculated UV transitions of the resveratrol–cyanidin complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 9.
MO and calculated UV transitions of the resveratrol–cyanidin complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 10.
MO and remaining of the calculated UV transitions of the resveratrol–cyanidin complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 10.
MO and remaining of the calculated UV transitions of the resveratrol–cyanidin complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 11.
MO and calculated UV transitions of the resveratrol–cyanidin–resveratrol complex at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 11.
MO and calculated UV transitions of the resveratrol–cyanidin–resveratrol complex at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 12.
MO and remaining of the calculated UV transitions of the resveratrol–cyanidin–resveratrol complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 12.
MO and remaining of the calculated UV transitions of the resveratrol–cyanidin–resveratrol complex at the ωB97X-D/6-31g+(d,p) theory level.
Figure 13.
Complex: resveratrol–cyanidin–resveratrol. UV spectrum with the three strongest transitions calculated. λmax 474.91 nm at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 13.
Complex: resveratrol–cyanidin–resveratrol. UV spectrum with the three strongest transitions calculated. λmax 474.91 nm at the ωB97X-D/6-31g+(d,p) level of theory.
Figure 14.
Hydrogen bonds analyzed with IGMPLOT software (version 3.08): hydrogen (26) joined to oxygen 46; hydrogen 61 joined to oxygen 31.
Figure 14.
Hydrogen bonds analyzed with IGMPLOT software (version 3.08): hydrogen (26) joined to oxygen 46; hydrogen 61 joined to oxygen 31.
Scheme 2.
Thermodynamic cycle for the formation of the copigmentation complex in water. The P and CoP symbols stand for pigment and copigment structures, respectively.
Scheme 2.
Thermodynamic cycle for the formation of the copigmentation complex in water. The P and CoP symbols stand for pigment and copigment structures, respectively.
Table 1.
Energy calculation, in Hartree, for the different species.
Table 1.
Energy calculation, in Hartree, for the different species.
| Theory Level | Cyanidin (Ha) | Resveratrol (Ha) | Complex (Ha) |
|---|
| M06-2x/6-31g+(d,p) (gd3bj) | Gas | −1028.975170 | −766.100919 | −1795.123485 |
| Water | −1029.050245 | −766.119796 | −1795.200952 |
| ωB97X-D/6-31g+(d,p) | Gas | −1029.043522 | −766.164797 | −1795.252825 |
| Water | −1029.118607 | −766.183451 | −1795.331088 |
| APFD/6-31g+(d,p) | Gas | −1028.580913 | −765.804968 | −1794.439441 |
| Water | −1028.654870 | −765.823624 | −1794.516138 |
Table 2.
Calculation of the binding energy for the different species.
Table 2.
Calculation of the binding energy for the different species.
| Theory Level | Without BSSE (kcal/mol) | With BSSE (kcal/mol) |
|---|
| M06-2x/6-31g+(d,p) (d3bj) | −29.74 | −25.77 |
| ωB97X-D/6-31g+(d,p) | −27.93 | −24.49 |
| APFD/6-31g+(d,p) | −33.61 | −29.46 |
Table 3.
DFT thermodynamic calculations at the theoretical level M06-2x/6-31g+(d,p) (gd3bj).
Table 3.
DFT thermodynamic calculations at the theoretical level M06-2x/6-31g+(d,p) (gd3bj).
| Theory Level | GAS | Cyanidin (Ha) | Resveratrol (Ha) | Complex (Ha) | kcal/mol * |
|---|
| M06-2x/6-31g+(d,p) (gd3bj) | Enthalpy | −1028.717780 | −765.855831 | −1794.617818 | −27.74 |
| Free energy | −1028.778627 | −765.914905 | −1794.715293 | −13.65 |
| Entropy | = −14.09 kcal/mol |
Table 4.
DFT thermodynamic calculations at the theoretical level of ωB97X-D/6-31+(d,p).
Table 4.
DFT thermodynamic calculations at the theoretical level of ωB97X-D/6-31+(d,p).
| Theory Level | GAS | Cyanidin (Ha) | Resveratrol (Ha) | Complex (Ha) | kcal/mol * |
|---|
| ωB97X-D/6-31g+(d,p) | Enthalpy | −1028.785195 | −765.919004 | −1794.745204 | −25.78 |
| Free energy | −1028.845790 | −765.979750 | −1794.843699 | −11.39 |
| Entropy | = −14.39 kcal/mol |
Table 5.
DFT thermodynamic calculations at the theoretical level of APFD/6-31+(d,p).
Table 5.
DFT thermodynamic calculations at the theoretical level of APFD/6-31+(d,p).
| Theory Level | GAS | Cyanidin (Ha) | Resveratrol (Ha) | Complex (Ha) | kcal/mol * |
|---|
| APFD/6-31g+(d,p) | Enthalpy | −1028.324304 | −765.560511 | −1793.934694 | −31.30 |
| Free energy | −1028.385209 | −765.619971 | −1794.032092 | −16.89 |
| Entropy | = −14.41 kcal/mol |
Table 6.
Binding free energy calculation.
Table 6.
Binding free energy calculation.
| Theory Level | Calculation According to Table 3, Table 4 and Table 5 |
|---|
| M06-2x/6-31g+(d,p) (gd3bj) | −13.65 + 10.34 = −3.31 kcal/mol |
| ωB97X-D/6-31g+(d,p) | −11.39 + 9.71 = −1.68 kcal/mol |
| APFD/6-31g+(d,p) | −16.89 + 9.98 = −6.91 kcal/mol |
Table 7.
Excitation energies and oscillator strengths for the allowed transitions for the cyanidin optimized energy, E(TD-HF/TD-DFT) = −1028.944 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
Table 7.
Excitation energies and oscillator strengths for the allowed transitions for the cyanidin optimized energy, E(TD-HF/TD-DFT) = −1028.944 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
| Excited State 1: | Singlet-A | 2.6107 eV | 474.91 nm | f = 0.5722 |
| 73- > 75 | −0.16981 | | | |
| 74- > 75 | 0.67064 | | | |
| Excited State 2: | Singlet-A | 3.0797 eV | 402.58 nm | f = 0.0189 |
| 73- > 75 | 0.66708 | | | |
| 74- > 75 | 0.17063 | | | |
| Excited State 3: | Singlet-A | 3.6213 eV | 342.38 nm | f = 0.1877 |
| 71- > 75 | 0.10239 | | | |
| 72- > 75 | 0.66697 | | | |
| 73- > 76 | 0.10369 | | | |
| Orbital | Energy (a.u.) | Contribution per simple atomic orbitals |
| 71 | −12.933 | 0.17 (C 1-1pz) 0.09 (C 1-2pz) 0.08 (O 27-1pz) 0.07 (C 16-1pz) 0.05 (O 25-1pz) |
| 72 | −12.203 | 0.10 (C 16-1pz) 0.09 (C 5-1pz) 0.08 (C 15-1pz) 0.07 (C 1-1pz) |
| 73 | −11.570 | 0.09 (C 15-1pz) 0.08 (C 5-1pz) 0.08 (O 31-1pz) |
74
HOMO | −10.823 | 0.08 (C 20-1pz) 0.07 (O 31-1pz) 0.07 (C 18-1pz) 0.06 (C 13-1pz) 0.06 (O 29-1pz) 0.06 (C 14-1pz) |
75
LUMO | −5.091 | 0.12 (C 11-1pz) 0.11 (C 9-1pz) 0.10 (C 9-2pz) 0.09 (C 11-2pz) 0.06 (O 22-1pz) 0.06 (C 9-3pz) |
| 76 | −2.694 | 0.09 (C 12-2pz) 0.07 (C 4-2pz) 0.07 (C 12-1pz) 0.06 (C 1-3pz) 0.06 (C 4-1pz) 0.05 (C 6-2pz) 0.05 (C 12-3pz) |
Table 8.
Excitation energies and oscillator strengths for the allowed transitions for the resveratrol. Optimized energy, E(TD-HF/TD-DFT) = −66.022 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
Table 8.
Excitation energies and oscillator strengths for the allowed transitions for the resveratrol. Optimized energy, E(TD-HF/TD-DFT) = −66.022 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
| Excited state 1: | Singlet-A | 3.6088 eV | 343.56 nm | f = 0.9407 |
| 60- > 61 | 0.69394 | | HOMO-LUMO |
| Excited State 2: | Singlet-A | 4.3685 eV | 283.82 nm | f = 0.0361 |
| 59- > 61 | 0.62744 | | | |
| 59- > 66 | −0.14339 | | | |
| 60- > 68 | −0.12492 | | | |
| 60- > 69 | −0.10503 | | | |
| Excited State 3: | Singlet-A | 4.5685 eV | 271.39 nm | f = 0.0744 |
| 57- > 61 | −0.25780 | | | |
| 58- > 62 | −0.19158 | | | |
| 60- > 62 | 0.59658 | | | |
| Orbital | Energy (a.u.) | Contribution per simple atomic orbitals |
| 57 | −9.397 | 0.16 (C 23-1pz) 0.15 (C 18-1pz) 0.14 (C 21-1pz) 0.13 (C 20-1pz) 0.07 (C 23-2pz) 0.06 (C 21-2pz) 0.06 (C 18-2pz) 0.06 (C 20-2pz) |
| 58 | −8.823 | 0.20 (C 8-4s) 0.12 (C 23-4s) 0.09 (C 18-4s) 0.09 (C 6-4s) 0.07 (C 19-4s) 0.07 (C 1-3px) 0.06 (C 3-3px) 0.05 (C 5-4s) |
| 59 | −8.124 | 0.12 (C 7-1pz) 0.11 (C 6-1pz) 0.07 (C 7-2pz) 0.06 (O 14-1pz) 0.06 (C 20-4s) 0.06 (O 16-1pz) 0.06 (C 6-2pz) 0.06 (C 10-1pz) 0.06 (C 8-1pz) |
| 60 | −6.962 | 0.19 (C 8-4s) 0.13 (C 7-4s) 0.11 (C 20-4s) 0.07 (C 10-4s) |
| 61 | −0.313 | 0.23 (C 23-4s) 0.10 (C 18-4s) 0.08 (C 18-3px) 0.06 (C 7-4s) 0.05 (C 8-4s) |
Table 9.
Excitation energies and oscillator strengths for the allowed transitions for the cyanidin–resveratrol complex. Optimized energy, E(TD-HF/TD-DFT) = −795.1849 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
Table 9.
Excitation energies and oscillator strengths for the allowed transitions for the cyanidin–resveratrol complex. Optimized energy, E(TD-HF/TD-DFT) = −795.1849 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
| Excited State 1: | Singlet-A | 1.3461 eV | 921.04 nm | f = 0.0040 |
| 134- > 135 | 0.70036 | | | |
| Excited State 2: | Singlet-A | 2.6446 eV | 468.83 nm | f = 0.2707 |
| 129- > 135 | 0.12937 | | | |
| 130- > 135 | 0.23485 | | | |
| 131- > 135 | 0.11971 | | | |
| 132- > 135 | 0.49142 | | | |
| 133- > 135 | 0.3821 | | | |
| Excited State 3: | Singlet-A | 3.0193 eV | 410.64 nm | f = 0.0577 |
| 131- > 135 | −0.15037 | | | |
| 132- > 135 | −0.41081 | | | |
| 133- > 135 | 0.53047 | | | |
| Orbital | Energy (a.u.) | Contribution per simple atomic orbitals |
| 129 | −11.685 | 0.08 (C11-4s) 0.07 (C54-4s) |
| 130 | −11.553 | 0.08 (C6-4s) 0.07 (C16-4s) 0.07 (C14-4s) 0.06 (C54-4s) |
| 131 | −11.147 | 0.10 (C6-4s) 0.09 (C11-4s) 0.06 (C37-4s) 0.06 (C40-4s) 0.05 (C14-4s) |
132
HOMO | −10.939 | 0.09 (C4-4s) 0.06 (C12-4s) 0.06 (C20-4s) 0.06 (C18-4s) 0.05 (C40-4s) 0.05 (C50-4s) |
| 133 | −10.476 | 0.14 (C6-4s) 0.13 (C2-4s) 0.09 (C14-4s) 0.07 (C5-4s) 0.06 (C38-4s) |
| 134 | −9.392 | 0.12 (C4-4s) 0.09 (C12-4s) 0.06 (C38-4s) 0.05 (C9-4s) |
135
LUMO | −4.950 | 0.12 (C50-4s) 0.12 (C52-4s) 0.09 (C37-4s) 0.07 (C55-4s) |
Table 10.
Excitation energies and oscillator strengths for the allowed transitions for the resveratrol–cyanidin–resveratrol complex. Optimized energy, E(TD-HF/TD-DFT) = −561.3871 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
Table 10.
Excitation energies and oscillator strengths for the allowed transitions for the resveratrol–cyanidin–resveratrol complex. Optimized energy, E(TD-HF/TD-DFT) = −561.3871 Ha. Level of theory: ωB97X-D/6-31g+(d,p) (f = oscillator strength).
| Excited state 1: | Singlet-A | 1.3922 eV | 890.59 nm | f = 0.0046 |
| 194- > 195 | 0.69822 | | | |
| Excited State 2: | Singlet-A | 2.2697 eV | 546.26 nm | f = 0.0090 |
| 193- > 195 | 0.70016 | | | |
| Excited State 3: | Singlet-A | 2.5730 eV | 481.87 nm | f = 0.2045 |
| 187- > 195 | 0.1405 | | | |
| 188- > 195 | −0.17338 | | | |
| 189- > 195 | −0.12092 | | | |
| 191- > 195 | 0.49147 | | | |
| 192- > 195 | −0.39274 | | | |
| Orbital | Energy (a.u.) | Contribution per simple atomic orbitals |
| 190 | −10.765 | 0.14 (C6-4s) |
191
HOMO | −10.540 | 0.20 (C6-4s) 0.05 (C50-4s) |
| 192 | −10.085 | 0.22 (C6-4s) 0.07 (C5-4s) |
| 193 | −9.888 | 0.13 (C1-4s) 0.09 (C3-3px) 0.08 (C6-4s) 0.07 (C9-4s) |
194
LUMO | −9.146 | 0.07 (C4-3px) 0.07 (C11-3px) |
| 195 | −4.619 | 0.11 (C50-4s) 0.08 (C81-4s) 0.08 (C52-4s) 0.06 (C54-4s) 0.06 (C 79-4s) |
Table 11.
HOMO–LUMO frontier orbital GAPS at ωB97X-D/6-31+g(d,p).
Table 11.
HOMO–LUMO frontier orbital GAPS at ωB97X-D/6-31+g(d,p).
| System | Frontier Orbitals Energies (a.u.) |
|---|
| | HOMO | LUMO | ΔLM * |
|---|
| Cyanidin | −10.823 | −5.091 | 5.732 |
| Resveratrol | −6.962 | −0.313 | 6.649 |
| Cyanidin–Resveratrol | −9.392 | −4.950 | 4.442 & |
| Resveratrol–Cyanidin–Resveratrol | −9.146 | −4.619 | 4.527 & |
Table 12.
Quantification of interactions for the case of ωB97X-D/6-31g+(d,p).
Table 12.
Quantification of interactions for the case of ωB97X-D/6-31g+(d,p).
| X-Y | d (Å) | IBSI | PDA (E-01) | Direction of Asymmetry |
|---|
| H26-O46 | 2.012 | 0.048 | 176.9 | H26 --- > O46 |
| H61-O31 | 2.837 | 0.004 | 123.5 | H61 ---- > O31 |
Table 13.
Quantification of interactions for the case of APFD/6-31g+(d,p).
Table 13.
Quantification of interactions for the case of APFD/6-31g+(d,p).
| X-Y | d (Å) | IBSI | PDA (E-01) | Direction of Asymmetry |
|---|
| H26-O46 | 1.973 | 0.050 | 180.0 | H26 ---- > O46 |
| H61-O31 | 2.679 | 0.005 | 131.3 | H61 ---- > O31 |
Table 14.
Quantification of interactions for the case of M06-2X/6-31g + (d,p).
Table 14.
Quantification of interactions for the case of M06-2X/6-31g + (d,p).
| X-Y | d (Å) | IBSI | PDA (E-01) | Direction of Asymmetry |
|---|
| H26-O46 | 2.072 | 0.035 | 171.0 | H26 ---- > O46 |
| H61-O31 | 2.627 | 0.006 | 134.0 | H61 ---- > O31 |
Table 15.
Shannon aromaticity index calculations.
Table 15.
Shannon aromaticity index calculations.

 ,
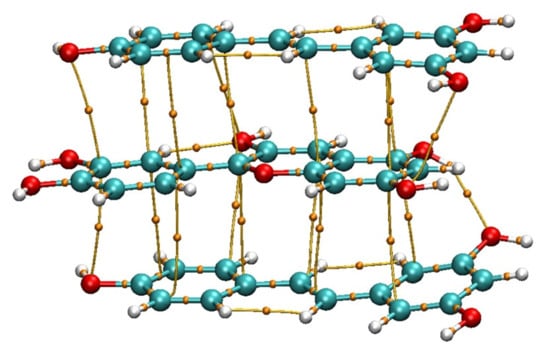
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
