
Computational Modeling to Identify Drugs Targeting Metastatic Castration-Resistant Prostate Cancer Characterized by Heightened Glycolysis

 , ,
, ,
Abstract
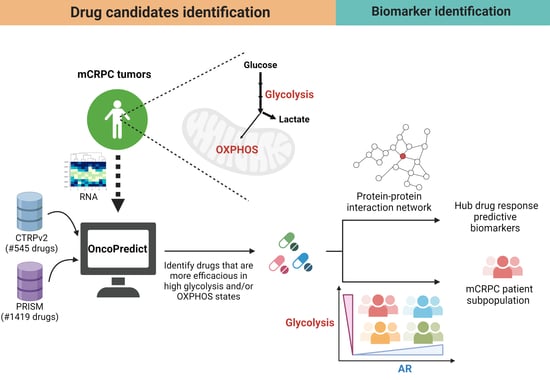
:
1. Introduction
2. Results
2.1. Identifying Candidate Drugs for mCRPC with High Glycolysis
2.2. Experimental Validation
2.3. Candidate Drug Biomarker Discovery
mCRPC Patients with Low AR Expression and High Glycolysis Were Predicted to Be More Sensitive to Ivemectin and P276-00
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Preprocessing
4.2. Impute Drug Response in Patients with mCRPC
4.3. Regression Analysis to Identify Efficacious Drugs for mCRPC with High Glycolysis
4.4. Selecting Drug Candidates with Higher Robustness for Validation
4.5. Cell Culture and Reagents
4.6. Cell Growth and Viability
4.7. Seahorse XFp Glycolysis Stress Test Assay
4.8. Stratifying Patients Based on AR and Glycolysis Status for Biomarker Discovery
4.9. Biomarker Discovery through PPI Network
4.10. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rebecca, L.; Siegel, K.D.M.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Chetta, P.; Sriram, R.; Zadra, G. Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications? Cancers 2023, 15, 3473. [Google Scholar] [CrossRef]
- Cui, Y.; Nadiminty, N.; Liu, C.; Lou, W.; Schwartz, C.T.; Gao, A.C. Upregulation of glucose metabolism by NF-kappaB2/p52 mediates enzalutamide resistance in castration-resistant prostate cancer cells. Endocr. Relat. Cancer 2014, 21, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.J.; Gavane, S.C.; Blanc-Autran, E.; Nehmeh, S.; Gonen, M.; Beattie, B.; Vargas, H.A.; Schoder, H.; Humm, J.L.; Fine, S.W.; et al. Positron Emission Tomography/Computed Tomography-Based Assessments of Androgen Receptor Expression and Glycolytic Activity as a Prognostic Biomarker for Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M.; Sankaranarayanan, M. A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. Eur. J. Med. Chem. 2020, 195, 112275. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla Rodriguez, B.S.; Correa, R. Raloxifene; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Guo, C.G.; Ma, W.; Drew, D.A.; Cao, Y.; Nguyen, L.H.; Joshi, A.D.; Ng, K.; Ogino, S.; Meyerhardt, J.A.; Song, M.; et al. Aspirin Use and Risk of Colorectal Cancer among Older Adults. JAMA Oncol 2021, 7, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; Eckert, S.; Krueger, K.A.; Grady, D.; Powles, T.J.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999, 281, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.J.; Crispin-Ortuzar, M.; Chin, S.F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, C.M.; Lamers, I.; Feys, P.; Gassert, R.; Lambercy, O. Personalized prediction of rehabilitation outcomes in multiple sclerosis: A proof-of-concept using clinical data, digital health metrics, and machine learning. Med. Biol. Eng. Comput. 2022, 60, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Saiful Islam, S.M.; Quinn, J.M.W.; Huq, F.; Moni, M.A. Machine learning and bioinformatics models to identify gene expression patterns of ovarian cancer associated with disease progression and mortality. J. Biomed. Inform. 2019, 100, 103313. [Google Scholar] [CrossRef] [PubMed]
- Severson, K.A.; Chahine, L.M.; Smolensky, L.A.; Dhuliawala, M.; Frasier, M.; Ng, K.; Ghosh, S.; Hu, J. Discovery of Parkinson’s disease states and disease progression modelling: A longitudinal data study using machine learning. Lancet Digit. Health 2021, 3, e555–e564. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Li, J.; Fu, Y.; Qu, L.; Tan, Y.; Zhang, P. A novel machine learning model based on sparse structure learning with adaptive graph regularization for predicting drug side effects. J. Biomed. Inform. 2022, 132, 104131. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Spisak, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O’Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 2021, 12, 5203. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Kumar, V.; Tailor, D.; Garcia-Marques, F.J.; Hsu, E.C.; Liu, S.; Bermudez, A.; Kanchustambham, V.; Shankar, V.; Inde, Z.; et al. SU086, an inhibitor of HSP90, impairs glycolysis and represents a treatment strategy for advanced prostate cancer. Cell Rep. Med. 2022, 3, 100502. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Kikuchi, R.; Kawagoe, J.; Tsuji, T.; Koyama, N.; Yamaguchi, K.; Nakamura, H.; Aoshiba, K. Anti-cancer strategy targeting the energy metabolism of tumor cells surviving a low-nutrient acidic microenvironment. Mol. Metab. 2020, 42, 101093. [Google Scholar] [CrossRef]
- Takatani-Nakase, T.; Matsui, C.; Maeda, S.; Kawahara, S.; Takahashi, K. High glucose level promotes migration behavior of breast cancer cells through zinc and its transporters. PLoS ONE 2014, 9, e90136. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [PubMed]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Adam, G.; Rampasek, L.; Safikhani, Z.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Machine learning approaches to drug response prediction: Challenges and recent progress. NPJ Precis. Oncol. 2020, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Shen, B.; Mou, X.; Li, Y.; Li, H. Large-scale pharmacogenomic studies and drug response prediction for personalized cancer medicine. J. Genet. Genomics 2021, 48, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, L.; Tang, Y.; Xie, D.; Wu, K.; Wei, W.; Xiao, Q. CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Sci. 2018, 109, 2590–2598. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Andrysik, Z.; Pandey, A.; Hoh, M.; Bonner, E.A.; Hill, A.A.; Sullivan, K.D.; Espinosa, J.M. CDK8 Kinase Activity Promotes Glycolysis. Cell Rep. 2017, 21, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.; Schcolnik-Cabrera, A.; Duenas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar] [PubMed]
- Pouliot, J.F.; L’Heureux, F.; Liu, Z.; Prichard, R.K.; Georges, E. Reversal of P-glycoprotein-associated multidrug resistance by ivermectin. Biochem. Pharmacol. 1997, 53, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, T.; Zhu, Z.; Hong, F.; Xu, Y.; Zhang, X.; Xu, X.; Ma, A. Ivermectin Augments the In Vitro and In Vivo Efficacy of Cisplatin in Epithelial Ovarian Cancer by Suppressing Akt/mTOR Signaling. Am. J. Med. Sci. 2020, 359, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, H.; Ikeda, A.; Takahashi, R.; Endo, H.; Sugawara, Y.; Goto, M.; Kanno, M.; Ogawa, S.; Nakamura, K.; Ujiie, H.; et al. Ivermectin represses Wnt/beta-catenin signaling by binding to TELO2, a regulator of phosphatidylinositol 3-kinase-related kinases. iScience 2022, 25, 103912. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Hu, X.; Wang, Y.; Yao, X.; Zhang, W.; Yu, C.; Cheng, F.; Li, J.; Fang, Q. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol. Res. 2021, 163, 105207. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, J.; Cai, B.; Bai, X.; Zhu, Y. Ivermectin accelerates autophagic death of glioma cells by inhibiting glycolysis through blocking GLUT4 mediated JAK/STAT signaling pathway activation. Environ. Toxicol. 2022, 37, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, Y.; Zhou, Z. Antibiotic ivermectin preferentially targets renal cancer through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 492, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Guler, E.M.; Akyildiz, A.G. Ivermectin Induces Oxidative Stress and DNA Damage in Breast Cancer Cells. Bezmialem Sci. 2023, 11, 15–22. [Google Scholar] [CrossRef]
- Lakshmana, G.; Baniahmad, A. Interference with the androgen receptor protein stability in therapy-resistant prostate cancer. Int. J. Cancer 2019, 144, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Farah, E.; Sarkar, S.; Ahmad, N.; Gupta, S.; Larner, J.; Liu, X. Cotargeting HSP90 and Its Client Proteins for Treatment of Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef]
- Boll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.K.; Heilbrun, L.K.; Sheng, S.; Stein, M.; Liu, G.; Antonarakis, E.S.; Vaishampayan, U.; Dzinic, S.H.; Li, X.; Freeman, S.; et al. A phase II trial of ganetespib, a heat shock protein 90 Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trials consortium (PCCTC) study. Investig. New Drugs 2016, 34, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 15. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Asberry, A.M.; Liu, S.; Nam, H.S.; Deng, X.; Wan, J.; Hu, C.D. Reprogramming landscape highlighted by dynamic transcriptomes in therapy-induced neuroendocrine differentiation. Comput. Struct. Biotechnol. J. 2022, 20, 5873–5885. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; Izumi, K.; Mizokami, A. Undesirable Status of Prostate Cancer Cells after Intensive Inhibition of AR Signaling: Post-AR Era of CRPC Treatment. Biomedicines 2021, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.C.; Ettinger, S.L.; Lin, D.; Xue, H.; Ci, X.; Nabavi, N.; Bell, R.H.; Mo, F.; Gout, P.W.; Fleshner, N.E.; et al. Targeting MCT4 to reduce lactic acid secretion and glycolysis for treatment of neuroendocrine prostate cancer. Cancer Med. 2018, 7, 3385–3392. [Google Scholar] [CrossRef] [PubMed]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome Biogenesis: A Central Player in Cancer Metastasis and Therapeutic Resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Mangangcha, I.R.; Malik, M.Z.; Kucuk, O.; Ali, S.; Singh, R.K.B. Identification of key regulators in prostate cancer from gene expression datasets of patients. Sci. Rep. 2019, 9, 16420. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, Z.; Wang, Y.; Xue, S. Identification of crucial genes and pathways associated with prostate cancer in multiple databases. J. Int. Med. Res. 2021, 49, 3000605211016624. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hu, Y.; Stearns, M.E. RPS2: A novel therapeutic target in prostate cancer. J. Exp. Clin. Cancer Res. 2009, 28, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Zhou, L.M.; Yuan, L.L.; Gao, Y.; Kui, X.Y.; Liu, X.Y.; Pei, Z.J. NPM1 Is a Prognostic Biomarker Involved in Immune Infiltration of Lung Adenocarcinoma and Associated with m6A Modification and Glycolysis. Front. Immunol. 2021, 12, 724741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.M.; Yuan, L.L.; Gao, Y.; Liu, X.S.; Dai, Q.; Yang, J.W.; Pei, Z.J. Nucleophosmin 1 overexpression correlates with (18)F-FDG PET/CT metabolic parameters and improves diagnostic accuracy in patients with lung adenocarcinoma. Eur. J. Nucl. Med. Mol Imaging 2021, 48, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shi, M.; Chen, H.; Gu, J.; Zhang, J.; Shen, B.; Deng, X.; Xie, J.; Zhan, X.; Peng, C. NPM1 activates metabolic changes by inhibiting FBP1 while promoting the tumorigenicity of pancreatic cancer cells. Oncotarget 2015, 6, 21443–21451. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Takeda, T.; Nakagawa, H.; Iwata, S.; Watanabe, T.; Siddiquey, M.N.; Goshima, F.; Murata, T.; Kawada, J.; Ito, Y.; et al. The heat shock protein 90 inhibitor BIIB021 suppresses the growth of T and natural killer cell lymphomas. Front. Microbiol. 2015, 6, 280. [Google Scholar] [CrossRef] [PubMed]
- Burrows, F.; Zhang, H.; Kamal, A. Hsp90 activation and cell cycle regulation. Cell Cycle 2004, 3, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Huang, K.; Chen, Z.; Hu, M.; Bai, Y.; Lin, S.; Du, H. Characterization of Glycolysis-Associated Molecules in the Tumor Microenvironment Revealed by Pan-Cancer Tissues and Lung Cancer Single Cell Data. Cancers 2020, 12, 1788. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhao, L.; Bi, J.; Guan, Q.; Qi, A.; Wei, Q.; He, M.; Wei, M.; Zhao, L. Glycolysis gene expression profilings screen for prognostic risk signature of hepatocellular carcinoma. Aging 2019, 11, 10861–10882. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Mao, Y.; Lu, B.; Zhou, G.; Wang, J. A hypoxia risk signature for the tumor immune microenvironment evaluation and prognosis prediction in acute myeloid leukemia. Sci. Rep. 2021, 11, 14657. [Google Scholar] [CrossRef]
- Li, W.; Huang, K.; Wen, F.; Cui, G.; Guo, H.; He, Z.; Zhao, S. Intermittent hypoxia-induced downregulation of microRNA-320b promotes lung cancer tumorigenesis by increasing CDT1 via USP37. Mol. Ther. Nucleic Acids 2021, 24, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.J.; Yang, Y.F.; Cheng, C.F.; Tu, Y.T.; Chen, Y.R.; Lee, M.C.; Tsai, K.W. Phosphofructokinase Platelet Overexpression Accelerated Colorectal Cancer Cell Growth and Motility. J. Cancer 2023, 14, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Du, Y.; Wang, L.; Chen, Z.; Liu, X. Weighted gene co-expression network analysis identifies CCNA2 as a treatment target of prostate cancer through inhibiting cell cycle. J. Cancer 2020, 11, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.L.; Zhang, W.; Lee, A.; Xia, Y.; Su, M.C.; Gruener, R.F.; Jena, S.; Huang, Y.; Pareek, S.; Shan, Y.; et al. Simplicity: Web-Based Visualization and Analysis of High-Throughput Cancer Cell Line Screens. J. Cancer Sci. Clin. Ther. 2023, 7, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Drugs Showing High Efficacy in High-Glycolysis mCRPC Patients (DrugHG) | ||||
| Clinical study | SU2C/EC | SU2C/WC | SU2C/EC | SU2C/WC |
| Training Drug database | CTRPv2 | CTRPv2 | PRISM | PRISM |
| Drugs predicted to show high efficacy in high-glycolysis mCRPC patients | 41 | 5 | 260 | 67 |
| DrugHG (SU2C/EC ∩ SU2C/WC) | 4 | 54 | ||
| DrugHG in primary MOA #,* | 0 | 9 & | ||
| (B) Dual-Effect Drugs (DrugDE) | ||||
| Clinical study | SU2C/EC | SU2C/WC | SU2C/EC | SU2C/WC |
| Training drug database | CTRPv2 | CTRPv2 | PRISM | PRISM |
| Drugs for high glycolysis mCRPC (G) | 41 | 5 | 260 | 67 |
| Drugs for high-OXPHOS mCRPC (O) | 80 | 59 | 530 | 492 |
| DrugDE (G ∩ O) | 13 | 0 | 156 | 27 |
| DrugDE (SU2C/EC ∩ SU2C/WC) | 0 | 19 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, M.-C.; Lee, A.M.; Zhang, W.; Maeser, D.; Gruener, R.F.; Deng, Y.; Huang, R.S. Computational Modeling to Identify Drugs Targeting Metastatic Castration-Resistant Prostate Cancer Characterized by Heightened Glycolysis. Pharmaceuticals 2024, 17, 569. https://doi.org/10.3390/ph17050569
Su M-C, Lee AM, Zhang W, Maeser D, Gruener RF, Deng Y, Huang RS. Computational Modeling to Identify Drugs Targeting Metastatic Castration-Resistant Prostate Cancer Characterized by Heightened Glycolysis. Pharmaceuticals. 2024; 17(5):569. https://doi.org/10.3390/ph17050569
Chicago/Turabian StyleSu, Mei-Chi, Adam M. Lee, Weijie Zhang, Danielle Maeser, Robert F. Gruener, Yibin Deng, and R. Stephanie Huang. 2024. "Computational Modeling to Identify Drugs Targeting Metastatic Castration-Resistant Prostate Cancer Characterized by Heightened Glycolysis" Pharmaceuticals 17, no. 5: 569. https://doi.org/10.3390/ph17050569