1. Introduction
In many areas, there is a great advantage in using living organisms or the components of living organisms (e.g., enzymes) as mild and selective biocatalysts instead of conventional chemical reactions for producing desired products. Biocatalysis offer safety and often enhances the economy and sustainability of reaction routes, in addition to the intrinsic stereo-, regio-, or enantioselectivity of the process [
1].
Using native or immobilized whole cells can be advantageous, as it avoids the need for costly enzyme purification processes, but whole-cell biotransformations suffer from mass transport issues, due to the cell wall acting as a barrier or competing enzymes within the cell, potentially causing side reactions [
2]. The use of isolated native enzyme may be advantageous to avoid these issues, but their production requires prior purification steps [
3].
For biotransformations with isolated enzymes, the enzyme produced within a cell must be separated from other cell components and further purified. Moreover, if the enzyme is applied in its native form, recovery in its active form from the reaction mixture may be difficult. Immobilizing enzymes onto proper carriers can simplify the recovery procedures and may provide enhanced stability and improved catalytic properties. In addition to enhanced recovery and reuse, enzyme immobilization can render enzymes more resistant to environmental influences (pH, temperature, solvent tolerance) [
4].
A variety of methods have been used to create immobilized enzymes as biocatalysts, ranging from enzyme entrapment in a matrix via binding to a carrier to cross-linking [
5,
6,
7,
8,
9]. Entrapment in a matrix is a simple, general, and efficient way to enhance stability, with the disadvantage of enhanced diffusion limitations within the matrix. There are many polymer matrices with different properties for biocatalyst entrapment to choose from [
10]. Enzymes can be immobilized on solid carriers as well. Several types of binding, such as adsorption, or attachment by ionic, covalent, or affinity interactions, have been used for immobilization onto various carriers, e.g., porous polymers, nanofibers, nanotubes, and nanoparticles [
11]. The advantage of covalent attachment to carriers is the stability of the binding, preventing leaching of the enzyme in response to external influences, and reducing diffusional limitations for substrate access and product release. With the right choice of the functional group for covalent binding, multipoint covalent attachment of the enzyme can be achieved, rendering the enzyme more stable on the surface, especially in the case of multimeric enzymes [
12].
Epoxy-functionalized solid carriers—offering the possibility of multipoint covalent binding—are frequently applied due to their stability and long-term storability. Due to their benefits, many of them are commercially available at reasonable prices. Several epoxy-functionalized carriers have been used, such as porous polymers [
13], silicates [
14], magnetic nanoparticles [
15], and even nanofibers [
16] or nanotubes [
17]. The main disadvantage of these covalent immobilization methods is the lack of selectivity. Thus, to achieve higher activity in epoxy-based immobilization, enzyme purification prior to use is needed.
To avoid the requirement of purification, a system offering the possibility of selective binding is needed. Affinity-based methods are the most suitable for providing selectivity. In 1975, Porath and colleagues discovered that proteins containing surface histidine and cysteine could coordinate with transition metals. They successfully separated serum proteins by coupling zinc and copper ions to an agarose-based resin modified with iminodiacetic acid [
18]. Later, Hochuli and co-workers purified polyhistidine-tagged proteins using this method [
19]. Today, Immobilized Metal Affinity Chromatography (IMAC), i.e., chromatographic separation based on the reversible binding of proteins to immobilized metal ions, has become one of the most widely used protein separation methods [
20]. In addition to the purification of histidine-tagged proteins, IMAC methods can also be applied for the single-step immobilization of enzymes with sufficiently high selectivity. The disadvantage of IMAC-type immobilization is the possible elution of attached enzymes from the surface by a stronger competitive complexing agent [
21,
22,
23].
The use of a bifunctional carrier providing a combination of selectivity function with a non-selective but stronger binding function offers the parallel advantages of the selectivity needed to avoid tedious purification and covalent binding to a carrier providing enhanced binding stability. One possibility is the use of IMAC-type affinity functions to create a bifunctional surface required for selective immobilization. Such metal affinity functions can be created by the modification of the support containing epoxy groups with iminodiacetic acid (IDA)- or nitrilotriacetic acid (NTA)-related functions. Iminodiacetic acid can react directly with epoxy groups on the surface to provide a weak chelating function [
23,
24,
25]. Nitrilotriacetic acid-like chelators with stronger metal chelation can be created by binding α-
N,N-dicarboxyalkylated
l-lysine derivatives through their free γ-amino function [
22,
26,
27,
28,
29,
30].
A convenient way of upgrading a cost-efficient but non-selective commercial epoxy-functionalized carrier is the easy-to-perform doping with various diamines first, followed by the creation of a metal-complexing function by treating the unreacted amino group of the linker diamine with a proper reagent that provides a chelator function and finally charging with a suitable metal ion. Various anhydrides, such as the ones from ethylenediaminetetraacetic acid (EDTADa), nitrilotriacetic acid (NTAa), or diethylenetriaminepentaacetic acid (DTPADa), allow the creation of different types of complexing agents on the surface.
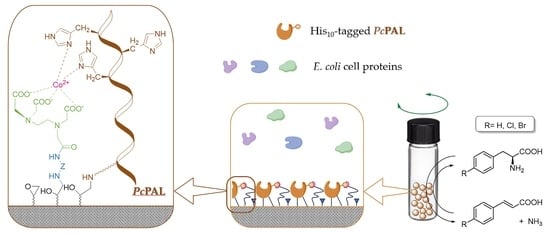
In this study, a commercially available macroporous, epoxy-functionalized poly(methyl methacrylate) carrier—the hydrophilic T2-150 resin—was used. Four different diamines with various length and properties were applied in sub-stoichiometric amounts relative to the epoxy group amounts, for partial surface modification. The unreacted amino group of the linker diamine was derivatized by EDTADa to create a metal ion complexing function, which was finally charged with Co
2+ ions (
Figure 1). The phenylalanine ammonia-lyase from
Petroselinum crispum (
PcPAL) [
31] was selected as the model enzyme for this case study. This biocatalyst can catalyze the reversible oxidative deamination of
l-phenylalanine to produce (
E)-cinnamic acid [
32], which is a precursor for the biosynthesis of lignin, several flavonoids, and phenylpropanoid compounds. The
PcPAL, carrying a polyhistidine affinity tag (His-tag), was produced recombinantly using
E. coli cells [
33].
2. Results and Discussion
This study intends to investigate the effect of the amount and quality of several diamines used as linkers for doping the surface of a cost-efficient epoxy carrier with a metal affinity binding function, on the selectivity, binding capacity, and activity of the selected phenylalanine ammonia-lyase from parsley (
PcPAL) as a test enzyme. The macroporous hydrophilic epoxy resin T2-150 from ChiralVision, representing cost efficient commercial epoxy carriers, was selected as the basic immobilization support. Due to its hydrophilic surface, it can be used conveniently as an enzyme immobilization support for reactions in an aqueous medium, and the 150–300 μm diameter of the beads renders them applicable in batch reactions as well as in packed bed reactors. Surface doping tests were performed with four diamine linkers of different lengths and flexibility (
Figure 1). Based on our previous studies on the metal affinity immobilization of
PcPAL [
34], the metal ion complexing function was created by reacting the residual amin of the linker with EDTADa, followed by charging with cobalt(II) as the metal ion.
The ammonia elimination from
l-phenylalanine (
l-
1a) catalyzed by
PcPAL was selected as the test reaction during the optimization of enzyme immobilization. The performance of the optimized immobilized
PcPAL forms was further demonstrated by the enantiomer-selective ammonia eliminations from racemic phenylalanines (±)-
1a-c (
Figure 2a) and the enantiotope-selective ammonia addition reaction onto the corresponding (
E)-arylacrylates (
E)-
2a-c, resulting in the formation of enantiopure
l-phenylalanines
l-
1a-c (
Figure 2b).
2.1. Optimization of the Doping Densities of Metal Ion Complexing Groups
At first, epoxy titration was used to determine the epoxy group content of the selected T2-150 resin (2.59 ± 0.11 mmol/g;
Supplementary Information S3.2). Next, to create a broad range of doping densities of metal ion complexing groups on the surface of the epoxy resin, the surface was modified with a proper diamine at various diamine-to-support ratios, enabling the creation of the metal complexing function in different ratios relative to the residual epoxy groups. The effect of the diamine linker-to-support ratio on the final immobilization yield and activity of the immobilized
PcPAL (immobilized from a crude cell lysate) was optimized with ethylenediamine (E) as the simplest diamine linker for carrying the metal ion complexing groups (
Figure 3). Due to the short chain of ethylenediamine, only one of the two amino groups could react with the epoxy groups on the surface. The ethylenediamine/support ratio in the diamine modification step was varied (ranging from 0.01 to 100 mmol g
−1 in 12 different preparations), followed by the creation of the metal ion complexing function with EDTADa derivatization and final charging with cobalt(II) ions. To ensure the reproducibility of the procedure, the primary amino group content of the diamine-modified resins was measured in each case (
Supplementary information, Table S2). In the chelator-forming step, two equivalents of EDTADa—relative to the measured amino group content—were applied.
After charging the created metal ion complexing function on the modified resins with cobalt(II) ions, the immobilization of polyhistidine(His
10)-tagged
PcPAL (produced by expression in recombinant
E. coli cells) was performed from crude cell lysate after separating the cell debris with centrifugation. Two washing protocols of the resins after the
PcPAL-loading step were used to determine the extent of
PcPAL binding only by complex formation and the proportion of
PcPAL attached by covalent bonds. When the enzyme-loaded resins were washed with salt solutions only, the weakly and non-selectively adsorbed proteins were removed only. Molecules attached by metal chelation or even stronger covalent bonds remained attached to the carrier. When a high-concentration imidazole solution was applied after washing with salt buffers, the fraction of
PcPAL retained only by metal complex formation were also eluted, and only the fraction of the enzyme remained on the resin attached covalently to the surface. The biocatalytic activity of the various forms of
PcPAL biocatalysts produced in these ways was tested in the ammonia elimination reaction of
l-
1a (
Figure 3).
The analysis of the full immobilization yield (
Yi) and specific activity (
UB) after the selective binding indicated that a sufficient complexation capacity (binding > 90% of
PcPAL with
UB ~15 U g
−1) could be reached at a 0.3 mmol g
−1 diamine/support ratio during the surface modification (
Figure 3a). Above this value, neither the total immobilization yield (
Yi) nor the specific activity (
UB) increased further significantly.
The immobilization yield (
Yic) and specific activity (
UB) of the covalently attached fraction of
PcPAL were analyzed in another series of immobilizations of the His-tagged
PcPAL by metal-affinity-function-doped epoxy resins, after implementing a further washing step with a high imidazole concentration solution after the selective binding to remove the non-covalently bound fraction of the enzyme. After the removal of the non-covalently attached fraction of
PcPAL from the surface, optimal values for the covalent immobilization yield (at
Yic = 87%) and specific activity (at
UB = 10.3 U g
−1) were observed at a 0.3 mmol g
−1 diamine/support ratio (
Figure 3b). When the amount of ethylenediamine was increased, the amount of covalently bound
PcPAL decreased, resulting in a very low covalent immobilization yield (
Table S4). This was in line with the decreasing amount of epoxy groups capable of forming covalent bonds. Based on the total epoxy content and the determined amount of free primary amino groups at the 0.3 mmol g
−1 diamine/support ratio (0.177 μmol g
−1,
Supplementary Information S3.3), the epoxy:amino group ratio on the surface at the optima was around 14:1.
Because the unmodified epoxy carrier proved suitable for covalent enzyme immobilization without any modification,
PcPAL immobilization was performed by the original T2-150 resin for comparison. The
PcPAL immobilization on the T2-150 resin was carried out from the crude cell lysate (bars 0 in panels (a,b) in
Figure 3) and from a purified
PcPAL solution (bars 0
P in panels (a,b) in
Figure 3). The specific activities of the T2-150-
PcPALs (
UB = 1.8 U g
−1 from cell lysate, and
UB = 4.7 U g
−1 from purified
PcPAL solution) remained far below the one achievable with the optimal surface-modified selective carrier directly from the cell lysate (
UB = 10.2 U g
−1 of E
0.3-
PcPAL).
2.2. Effect of Various Diamines as a Linker Unit on the Performance of the Bifunctional Support
Based on the experiments with ethylenediamine, the optimum covalent binding ability of the resulting metal-affinity/epoxy bifunctional resins was assumed to be between 0.1–0.5 mmol g−1 diamine/support ratio for other diamines with terminal primary amine function providing similar reactivity. The surface modification protocol was employed to three additional diamines, namely diethylenetriamine (D), 2,2′-(ethylenedioxy)bis(ethylamine) (O), p-xylylenediamine (X). Two of these diamines (D and O) have longer chains than ethylenediamine (E), providing an even higher degree of flexibility. Due to the planar structure of the aromatic ring in p-xylylenediamine (X), this linker is less flexible but more hydrophobic than the others. Expectedly, the different nature of these linkers could affect the surface properties and thus the outcome of enzyme immobilization. To lower experimental efforts, these diamines (D, O, and X) were used only at five ratios around the previously found optimum with linker E (0.1, 0.2, 0.3, 0.4, 0.5 mmol g−1 diamine/support ratio).
In fact, only minor differences were observed between the affinity function-doping results with the different diamines (
Figure 4). In the case of all the three linear chain linker diamines (E, D, and O), the best specific activity of the covalently fixed
PcPAL was achieved at the 0.3 mmol g
−1 diamine/support ratio, while for the bifunctional supports obtained with the aromatic ring-containing diamine (X), the 0.2 mmol g
−1 diamine/support ratio was the best. However, when considering the resulting amount of primary amino groups introduced to the surface after diamine derivatization (
Supplementary Informations Table S3), the optima were at almost the same amino group content (between 0.09 and 0.18 mmol g
−1).
These results clearly demonstrate that the amount of amine functions—thus the final density of the metal-ion-affinity-functions—has a more significant influence on the specific activity of the covalently bound PcPAL than the quality of the linker from the diamine. Taking the cost–benefit analysis for diamines into consideration, the diethylenetriamine linker (D) at the optimal diamine support ratio was selected for further investigations with the PcPAL bound covalently to bifunctional carrier (D0.3-PcPAL).
2.3. Comparison with the Iminodiacetic Acid-Modified Supports Lacking Linker Unit
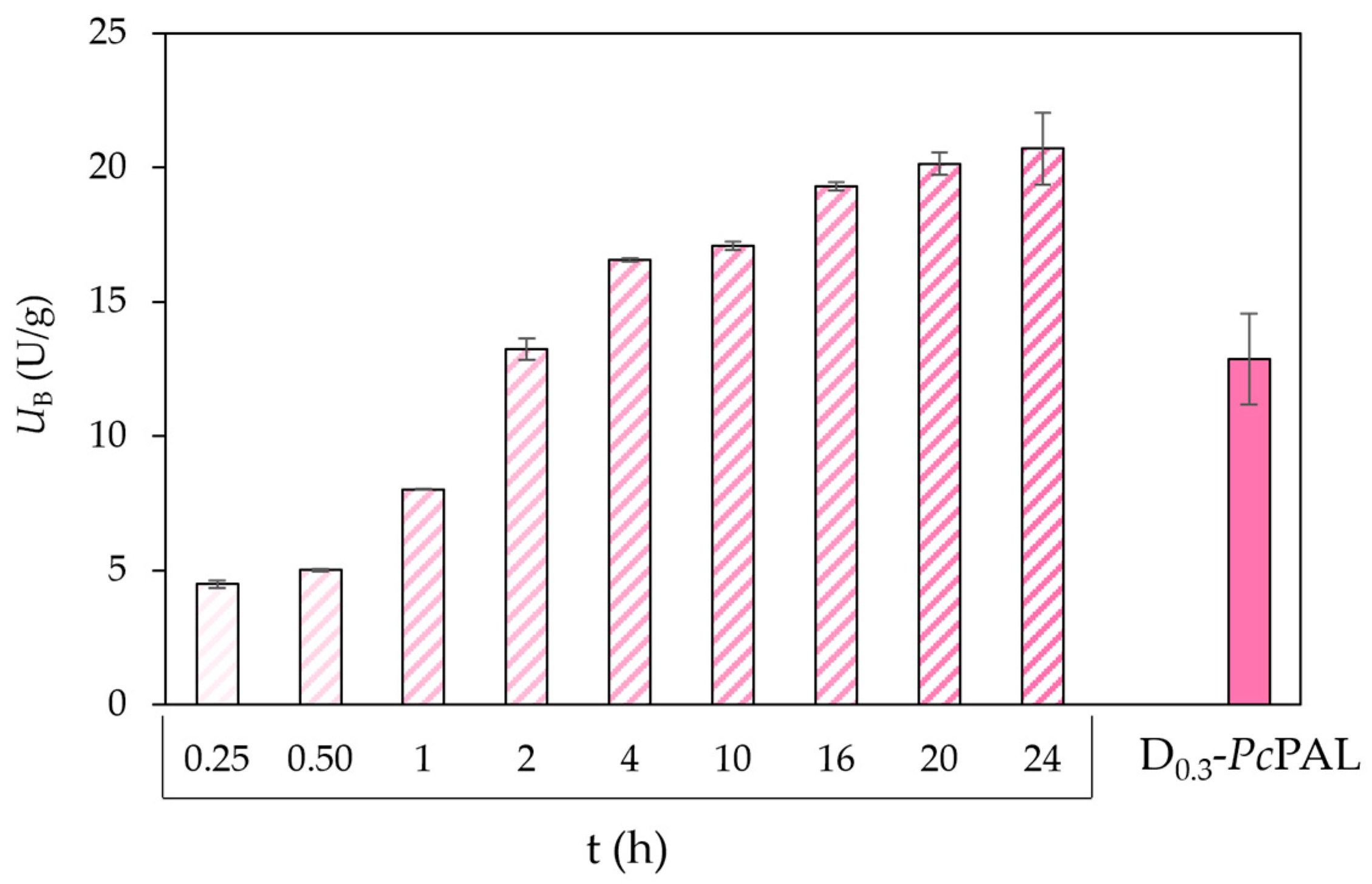
Bifunctional carriers with metal-ion-affinity-functions at epoxy-charged surface were most often created using iminodiacetic acid (IDA) [
35]. Sub-stoichiometric amounts of IDA could directly react with a part of the epoxy groups to form metal-ion-chelating units, and the residual epoxy groups were suitable for covalent bond formation. The amount of IDA introduced to the surface was typically controlled by the reaction time, rather than varying the reagent concentration. Mateo and co-workers found that lower densities of the forming metal-ion-chelating units on the surface were optimal in the selective immobilization of His-tagged enzymes.
As a comparison to the present bifunctional carriers with diamine-based linkers between the surface and the metal-ion-chelating units, the surface modification of the T2-150 epoxy carrier with IDA was performed in a similar way by varying the reaction time of IDA treatment between 15 min and 24 h. After loading
PcPAL-containing cell lysate onto the series of Co
2+-charged IDA-modified carriers, the non-covalently attached
PcPAL was eluted by washing with a high imidazole concentration solution, as in the previous studies on the
PcPAL-containing diamine linker-based bifunctional carriers. As shown in
Figure 5, the specific activity achieved with D
0.3-
PcPAL (
UB = 10.3 U g
−1) could be surpassed by the IDA-based carrier after the two-hour-long reaction. With longer surface modification times, the IDA
xh-
PcPAL biocatalysts provided even higher specific activities, up to
UB = 20.7 U g
−1 with the IDA
20h-
PcPAL. These results indicated a higher binding capacity of the IDA-based bifunctional carriers compared to the ones with more bulky metal-ion-chelating units attached to diamine linkers.
To compare the operational durability of the IDA-type
PcPAL to the ones with a diamine linker, the reusability of IDA
20h-
PcPAL and D
0.3-
PcPAL was studied in the ammonia elimination reaction from
l-
1a and in the ammonia addition reaction onto (
E)-
2a (
Figure 6). Under the milder conditions of the elimination reaction (phosphate buffer, pH = 8.8), a similar loss of activity was experienced with the two types of biocatalysts. However, in the ammonia addition reaction requiring much harsher reaction conditions (6 M ammonia solution), the D
0.3-
PcPAL—containing the diethylenetriamine-based linker and EDTA-derived metal-ion-affinity unit—clearly overperformed the operational stability of IDA
20h-
PcPAL. Thus, the biocatalysts on the diamine linker-based bifunctional carriers were more resistant to the demanding reaction conditions than the IDA-modified bifunctional epoxy supports.
2.4. Comparison of the PcPAL Immobilization on Non-Selective and Upgraded Epoxy Resins
The results could be rationalized by the absence of the selectivity for the epoxy resin which binds all the proteins from a mixture in ratio close to their statistical distribution (
Figure 7a). Since the unwanted binding of the protein impurities other than the target enzyme decrease the binding capacity of the non-selective epoxy resin, the immobilization yield as well as the specific activity are lowered in this case.
When immobilization is carried out on the parent epoxy resin from a pure enzyme solution, almost the entire amount of enzyme can be bound covalently onto the carrier. The observation of significantly lower specific activity for the enzyme immobilized on the parent epoxy carrier, as compared to the optimal bifunctional carrier, can be explained by the more oriented immobilization of the His-tagged enzyme. On the non-modified epoxy support, the enzyme can be bound covalently through any suitable reactive group (amino, thiol, etc.), adopting statistical orientations, including partially inactive ones due to hindering to the active center or entry tunnels for the substrates (
Figure 7b). In addition, multipoint attachments by covalent bonds close to the surface rigidify the enzyme, which may also result in activity decrease.
When parent epoxy resins are converted to metal-affinity-function-doped bifunctional carriers, the metal ion complexes ensure selectivity and orient the enzyme during covalent bond formation (
Figure 7c,d). Consequently, an active immobilized catalyst can be obtained from an His-tagged protein, even directly from a crude lysate. A slight difference can arise from the nature of the affinity function linker. If the affinity function is bound to the carrier surface without a linker (such as in the case of IDA doping,
Figure 7c), the orientation space of the enzyme may be limited, due to the limited availability of the functions for covalent bond formation at the enzyme surface. In the case of a longer linker (such as the diamine units in the present study) between the carrier surface and the affinity function in the bifunctional carrier (
Figure 7d), the orientation space of the enzyme increases, and due to more attachment points becoming available, the stability of enzyme binding is enhanced.
2.5. Scope of Applicability of the PcPAL Immobilized Selectively on the Upgraded Epoxy Resins
The performance, stability, and reusability of D0.3-PcPAL were characterized further in stereoselective biotransformations.
Ammonia elimination reactions from racemic α-phenylalanines (±)-
1a-c—including two halogenated non-natural amino acids (±)-
1b,c, in addition to the racemic α-phenylalanine (±)-
1a—were performed in five subsequent cycles (
Table 1). In these kinetic resolutions, the elimination took place from the
l-enantiomers
l-
1a-c, providing the corresponding €-cinnamic acids (
E)-
2a-c, while the corresponding
d-enantiomers
d-
1a-c remained unreacted. Although the activity continuously declined even during the elimination reactions, the catalyst retained at least 70% of its initial activity even in the fifth reaction cycle.
The opposite reaction, enantiotope-selective ammonia addition onto the achiral (E)-cinnamic acids (E)-2a-c, providing the enantiopure l-α-phenylalanines l-1a-c, was also carried out in five subsequent cycles. Quite high conversions were achieved even in the fifth reaction cycle, providing products l-1a-c with exclusive stereoselectivity. Under the harsh conditions of the amino acid synthesis (6M ammonia solution), the D0.3-PcPAL biocatalyst resisted the demanding conditions and could keep at least 70% of the original activity as well.
3. Materials and Methods
3.1. Materials
The macroporous hydrophilic polyacrylic epoxy resin T2-150 (D = 150–300 μm) was purchased from ChiralVision B.V. (Den Hoorn, Netherlands). Ethylenediaminetetraacetic acid (EDTA), potassium chloride, sodium chloride, tris(hydroxymethyl)aminomethane (TRIS), cobalt(II) acetate tetrahydrate, ethylenediamine, diethylenetriamine, 2,2′-(ethylenedioxy)bis(ethylamine),
p-xylylenediamine, (
E)-4-chlorocinnamic acid (
E)-
2b, and (
E)-4-bromocinnamic acid (
E)-
2c were purchased from Sigma-Aldrich (Saint Louis, MO, USA; now Merck KGaA, Darmstadt, Germany). Imidazole, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES),
l-phenylalanine
l-
1a, and (
E)-cinnamic acid (
E)-
2a were the products of Alfa Aesar Europe (Karlsruhe, Germany). Pyridine, acetic anhydride, and all solvents were purchased from Merck KGaA (Darmstadt, Germany). The technical-grade solvents were dried and/or freshly distilled prior to use, and dimethylformamide was dried over 4 Å molecular sieves. Substrates (±)-
1b,c were synthesized according to previously described methods [
36]. The method for the preparation of ethylenediaminetetraacetic anhydride (EDTADa) is given in the
Supplementary Information. The expression of recombinant phenylalanine ammonia-lyase from parsley (
PcPAL) carrying an
N-terminal His
10-tag in
E. coli Rosetta
TM host was carried out according to the method described by Dima et al. [
33] (
Supplementary Information Section S3.6).
3.2. Surface Treatment of T2-150 Carriers with Diamines
In a 20 mL screw cap vial, polymer beads (500 mg) were measured and suspended in 2-propanol (1 mL). The given amount of the specified diamine was added directly (in the case of 2.5; 5; 10; 20; 50; 100 mmol diamine/g polymer beads) or from a stock solution (200 mM in 2-propanol; in the case of 0.01; 0.05; 0.1;0.2; 0.3; 0.4; 0.5; 1 mmol diamine/g polymer beads), and the suspensions were completed with the addition of 2-propanol to a final volume of 5 mL. After shaking the resulting mixture at 450 rpm for 24 h at 60 °C, the resins were filtered off, washed with 2-propanol (3 × 10 mL), and dried in a vacuum drying chamber at room temperature for 5 h (until the vacuum level dropped below 10 m bar).
3.3. Derivatization of the Diamine-Doped T2-150 Carriers with EDTADa and Cobalt(II) Ions
In a dried 20 mL screw cap vial, the diamine-doped T2-150 beads (500 mg) were suspended in
N,
N-dimethylformamide (10 mL) with EDTADa (2 equivalents to the surface amino groups; for determination of amino group content, see
Section S3.2 in Supplementary Information) and
N,
N-diethyl-
N-isopropylamine (1 equivalent to the EDTADa). After shaking the resulting mixture at 450 rpm for 24 h at 60 °C, distilled water (500 µL) was added, and shaking was continued for a further hour at 60 °C. After this quenching, the resins were filtered off and washed with acetonitrile (2 × 10 mL), 2-propanol (10 mL), and distilled water (10 mL).
After shaking the derivatized resins in cobalt(II)-acetate solution (25 mL, 50 mM, in distilled water) in a 30 mL screw cap vial at 450 rpm for 30 min at room temperature, the metal-charged supports were filtered off and washed with distilled water (3 × 10 mL) and 2-propanol (10 mL), then dried in a vacuum drying chamber at room temperature for 5 h (until the vacuum level dropped below 10 mbar).
3.4. Immobilization of PcPAL on the T2-150 Epoxy Resin
In a 4 mL screw cap vial, the T2-150 epoxy resin (10 mg) was added to the solution of His-tagged PcPAL (2 mL, ~1 mg mL−1 purified PcPAL obtained by the standard Ni-NTA method; or the lysate of E. coli cells expressing His-tagged PcPAL, 2 mL, containing ~1 mg mL−1 of PcPAL) and the resulting suspension was shaken at 450 rpm for 16 h at 25 °C. The unbound proteins were washed with the following sequentially added solutions (2 mL each): low salt buffer (30 mM KCl; 50 mM HEPES, pH 7.5), high salt buffer (300 mM KCl, 50 mM HEPES, pH 7.5), high-concentration imidazole buffer (500 mM imidazole, 30 mM KCl, 50 mM HEPES, pH 7.5), and lysis buffer (50 mM TRIS, 150 mM NaCl; pH 8.0; 3 × 1 mL), and used directly in the test reactions.
The PcPAL activity of the PcPAL solutions was measured before and after the immobilization process. An aliquot of the corresponding PcPAL solution (20 μL) was added to the l-phenylalanine solution (l-1a, 1 mL, 10 mM in TRIS buffer (100 mM; pH 8.8)) at 30 °C, and the absorbance change after 5 min was detected at 290 nm.
A 5 mg sample of the immobilized PcPAL was added to the solution of l-phenylalanine l-1a (1 mL, 20 mM in 100 mM TRIS buffer; pH 8.8) in a 4 mL screw cap vial, and the mixture was shaken at 750 rpm at 30 °C. After 0.5, 1, 3, and 5 h, samples (20 µL) were taken and diluted with distilled water to a final volume of 300 μL.
The specific activity (UB) was calculated from the conversion from l-1a after 3 h reaction time (determined by UV-spectroscopy from the absorption change of the forming (E)-2a using the Lambert–Beer equation [at 290 nm, ε290=9790 L cm−1 mol−1; in a Thermo Scientific Multiscan SkyHigh microplate reader (Thermo Fisher Scientific Inc., Waltham, MA, USA) with 300 μL Greiner UV-Star® 96 well-plate)] using the equation UB = nP/(t × mB) (where nP [μmol] is the amount of the product, t [min] is the reaction time, and mB [g] is the mass of the applied biocatalyst). The measurement of PcPAL activities and determination of immobilization yield were performed according to Section S3.5 in the Supporting Information.
3.5. Selective Immobilization of PcPAL on the Chelator-Doped T2-150 Resins
In a 4 mL screw cap vial, the chelator-Co
2+-doped T2-150 epoxy resin (10 mg, with varied IDA-Co
2+ content, see
Figure 6; or with varied diamine-EDTADa-Co
2+, see
Figure 3 and
Figure 5) was added to the lysate of
E. coli cells expressing His-tagged
PcPAL (2 mL, containing ~1 mg mL
−1 PcPAL), and the resulting suspension was shaken at 450 rpm for 16 h at 25 °C. The unbound proteins were washed first with the sequential addition of the following solutions (2 mL, each): low salt buffer (30 mM KCl; 50 mM HEPES, pH 7.5), high salt buffer (300 mM KCl, 50 mM HEPES, pH 7.5) (see partial washing results of ethylendiamine-EDTADa-Co
2+resins in
Figure 3a). The non-covalently bound fraction of proteins was washed out by high-concentration imidazole buffer (500 mM imidazole, 30 mM KCl, 50 mM HEPES, pH 7.5) and lysis buffer (50 mM TRIS, 150 mM NaCl; pH 8.0; 3 × 1 mL) (for diamine-EDTADa-Co
2+resins, see
Figure 3b and
Figure 5; for IDA-Co
2+resins, see
Figure 6). The measurement of
PcPAL activities and determination of immobilization yield were performed according to Section S3.5 in the Supporting Information.
3.6. Comparison of the Reusability of PcPAL on Different Chelator-Doped T2-150 Resins
Reusability tests were performed in 4 mL screw cap vials using
PcPAL from a crude cell lysate on optimal chelator-Co
2+-doped T2-150 epoxy resins (5 mg, IDA
20h-
PcPAL, see
Figure 7a; or D
0.3-
PcPAL, see
Figure 7b) over five subsequent 4 h reaction cycles.
The recyclability in ammonia elimination started from an
l-phenylalanine
l-
1a solution (2 mL, 10 mM in 100 mM TRIS buffer; pH 8.8), shaken at 750 rpm at 30 °C for 4 h in five repeated cycles. After each cycle, the resins were washed (2 × 1 mL, 100 mM TRIS buffer; pH 8.8). From the reactions of each cycle, samples (20 µL) were taken at 1 h reaction time and diluted with distilled water to a final volume of 300 μL for activity determination, as described in
Section 3.4.
The recyclability in ammonia addition started from (
E)-cinnamate (
E)-
2a solution (1 mL, 5 mM in 6 M NH
3 solution; adjusted to pH 10.0 by the addition of dry ice), shaken at 750 rpm at 30 °C for 4 h in five repeated cycles. After each cycle, the resins were washed with ammonia solution (2 × 1 mL, 6 M NH
3 solution; adjusted to pH 10.0 by the addition of dry ice) and sodium phosphate buffer (1 mL, 50 mM, pH 6.5). From the reactions of each cycle, samples (20 µL) were taken at 1 h reaction time and diluted with distilled water to a final volume of 300 μL for activity determination, as described in
Section 3.4.
3.7. Comparison of the Operational Stability of PcPAL on Different Chelator-Doped T2-150 Resins in Reactions from Racemic Phenylalanines (±)-1a-c or (E)-2a-c
Operational stability tests for reactions from racemic phenylalanines (±)-
1a-c and from (
E)-
2a-c were performed in 4 mL screw cap vials using
PcPAL from a crude cell lysate on optimal chelator-Co
2+-doped T2-150 epoxy resins (5 mg, IDA
20h-
PcPAL, or D
0.3-
PcPAL; see
Table 1 over five subsequent 4 h reaction cycles (except for reactions from (±)-
1a, lasting only 3 h). Sampling was performed similarly as that described in
Section 3.6. Conversions were determined by UV-spectroscopy from the absorption change of
2a-c at 290 nm (
2a: ε
290 = 9790 L cm
−1 mol
−1;
2b: ε
290 = 14,617 L cm
−1 mol
−1;
2c: ε
290 = 17,594 L cm
−1 mol
−1). The enantiomeric excess of
l-1a-c and
d-
1a-c were determined with chiral HPLC at the end of every reaction cycle (see
Supplementary Information S2.1 and Figures S1–S3).
Testing the stability of the optimal chelator-Co2+-doped T2-150 epoxy resins (5 mg, IDA20h-PcPAL, or D0.3-PcPAL) in ammonia elimination reactions was performed using the solution of the corresponding racemic phenylalanine (±)-1a-c (2 mL; 10 mM (±)-1a or 5 mM for (±)-1b,c, in 100 mM TRIS buffer; pH 8.8), shaken at 750 rpm at 30 °C in five repeated cycles. After each cycle, the resins were washed (2 × 1 mL, 100 mM TRIS buffer; pH 8.8).
The recyclability of the optimal chelator-Co2+-doped T2-150 epoxy resins (5 mg, IDA20h-PcPAL, or D0.3-PcPAL) in ammonia addition reactions was performed using (E)-cinnamate (E)-2a-c solution (1 mL, 5 mM (E)-2a-c in 6 M NH3 solution; adjusted to pH 10.0 by the addition of dry ice), shaken at 750 rpm at 30 °C in five repeated cycles. After each cycle, the resins were washed with ammonia solution (2 × 1 mL, 6 M NH3 solution; adjusted to pH 10.0 by the addition of dry ice) and sodium phosphate buffer (1 mL, 50 mM, pH 6.5).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}