
Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes

 ,
,  ,
,  and
and
Abstract
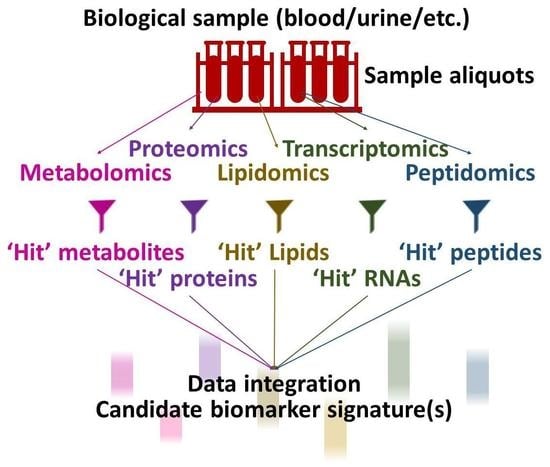
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Proteomics Analysis
2.3. Metabolomics Analysis
2.4. Lipidomics Analysis
2.5. Transcriptomics Analysis
2.6. Data Analysis
3. Results
3.1. Parallel Untargeted Quadra-Omics
3.2. The Impact of Thresholding Strategy on the Distribution of Features Identified by Quadra-Omics
3.3. Comparative Enrichment Analyses for Diseases and Functions, Canonical Pathways, and Upstream Regulators
3.4. Regulator Effects Analysis Predicting Cause-Effect Relationships in the Integrated Quadra-Omics Dataset
3.5. Integrative Molecular Network Analysis of Parallel Multi-Omics Datasets Facilitates the Identification of Potential T1D Biomarker Signature(s)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chast, F.; Slama, G. [Apollinaire Bouchardat and diabetes]. Hist. Sci. Med. 2007, 41, 287–301. [Google Scholar]
- Best, C.H. The internal secretion of the pancreas. J. Am. Med. Assoc. 1935, 105, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Bottazzo, G.F.; Florin-Christensen, A.; Doniach, D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974, 2, 1279–1283. [Google Scholar] [CrossRef]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomans de Brachène, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 2018, 36, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanou, M.; Protogerou, A.; Tsoucalas, G.; Androutsos, G.; Poulakou-Rebelakou, E. Milestones in the history of diabetes mellitus: The main contributors. World J. Diabetes 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghesquière, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An integrated view of immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [Green Version]
- Achenbach, P.; Bonifacio, E.; Koczwara, K.; Ziegler, A.G. Natural history of type 1 diabetes. Diabetes 2005, 54 (Suppl. 2), S25–S31. [Google Scholar] [CrossRef] [Green Version]
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, Å.; Rewers, M.J.; She, J.X.; Ziegler, A.G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A combined risk score enhances prediction of type 1 diabetes among susceptible children. Nat. Med. 2020, 26, 1247–1255. [Google Scholar] [CrossRef]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- Pugliese, A. The multiple origins of Type 1 diabetes. Diabet Med. 2013, 30, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Eisenbarth, G.S. Genetic control of autoimmunity in type I diabetes and associated disorders. Diabetologia 2002, 45, 605–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlquist, G.; Blom, L.; Holmgren, G.; Hägglöf, B.; Larsson, Y.; Sterky, G.; Wall, S. The epidemiology of diabetes in Swedish children 0-14 years—A six-year prospective study. Diabetologia 1985, 28, 802–808. [Google Scholar] [CrossRef]
- Wenzlau, J.M.; Hutton, J.C. Novel diabetes autoantibodies and prediction of type 1 diabetes. Curr. Diab. Rep. 2013, 13, 608–615. [Google Scholar] [CrossRef]
- Jacobsen, L.M.; Haller, M.J.; Schatz, D.A. Understanding pre-type 1 diabetes: The key to prevention. Front. Endocrinol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergholdt, R.; Brorsson, C.; Palleja, A.; Berchtold, L.A.; Fløyel, T.; Bang-Berthelsen, C.H.; Frederiksen, K.S.; Jensen, L.J.; Størling, J.; Pociot, F. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 2012, 61, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Onengut-Gumuscu, S.; Chen, W.-M.; Burren, O.; Cooper, N.J.; Quinlan, A.R.; Mychaleckyj, J.C.; Farber, E.; Bonnie, J.K.; Szpak, M.; Schofield, E.; et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015, 47, 381–386. [Google Scholar] [CrossRef]
- Nyaga, D.M.; Vickers, M.H.; Jefferies, C.; Perry, J.K.; O’Sullivan, J.M. The genetic architecture of type 1 diabetes mellitus. Mol. Cell Endocrinol. 2018, 477, 70–80. [Google Scholar] [CrossRef]
- Hopfgarten, J.; Stenwall, P.A.; Wiberg, A.; Anagandula, M.; Ingvast, S.; Rosenling, T.; Korsgren, O.; Skog, O. Gene expression analysis of human islets in a subject at onset of type 1 diabetes. Acta Diabetol. 2014, 51, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, A.M.; Mira, M.F.; Ebid, G.T.A.; Kassem, S.H.; Radwan, E.R.; Mamdouh, M.; Amin, M.; Badawy, N.; Bazaraa, H.; Ibrahim, A.; et al. Association of insulin gene VNTR INS -23/Hph1 A>T (rs689) polymorphism with type 1 diabetes mellitus in Egyptian children. Egypt. J. Med. Hum. Genet. 2019, 20, 13. [Google Scholar] [CrossRef] [Green Version]
- Vafiadis, P.; Bennett, S.T.; Todd, J.A.; Nadeau, J.; Grabs, R.; Goodyer, C.G.; Wickramasinghe, S.; Colle, E.; Polychronakos, C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 1997, 15, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.P.; Gillespie, K.M.; Thomson, G.; Cordell, H.J.; Todd, J.A.; Gale, E.A.; Bingley, P.J. Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: A population-based study in the United Kingdom. J. Clin. Endocrinol. Metab. 2004, 89, 4037–4043. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.M.; Gale, E.A.; Bingley, P.J. High familial risk and genetic susceptibility in early onset childhood diabetes. Diabetes 2002, 51, 210–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdi, A.M.; Hamilton-Williams, E.E.; Cristino, A.; Ziegler, A.; Bonifacio, E.; Le Cao, K.A.; Harris, M.; Thomas, R. A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Gastol, J.; Polus, A.; Biela, M.; Razny, U.; Pawlinski, L.; Solnica, B.; Kiec-Wilk, B. Specific gene expression in type 1 diabetic patients with and without cardiac autonomic neuropathy. Sci. Rep. 2020, 10, 5554. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yu, H.; Zhang, H.; Si, Y.; Tian, D.; Zhao, X.; Luan, J.; Jia, H. Integrated analysis of different microarray studies to identify candidate genes in type 1 diabetes. J. Diabetes 2017, 9, 149–157. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Li, R.; Chung, A.C.K.; Yu, X.; Lan, H.Y. MicroRNAs in diabetic kidney disease. Int. J. Endocrinol. 2014, 2014, 593956. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Vasu, S.; Kumano, K.; Darden, C.M.; Rahman, I.; Lawrence, M.C.; Naziruddin, B. microrna signatures as future biomarkers for diagnosis of diabetes states. Cells 2019, 8, 1533. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Zhang, X. The Profiling and Role of miRNAs in Diabetes Mellitus. J. Diabetes Clin. Res. 2019, 1, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Luk, A.O.Y.; Chan, J.C.N.; Ma, R.C.W. MicroRNA and diabetic complications: A clinical perspective. Antioxid. Redox Signal. 2017, 29, 1041–1063. [Google Scholar] [CrossRef]
- Scherm, M.G.; Daniel, C. miRNA Regulation of T cells in islet autoimmunity and type 1 diabetes. Curr. Diabetes Rep. 2020, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Ye, L.; Wang, B.; Gao, J.; Liu, R.; Hong, J.; Wang, W.; Gu, W.; Ning, G. Decreased miR-146 expression in peripheral blood mononuclear cells is correlated with ongoing islet autoimmunity in type 1 diabetes patients 1miR-146. J. Diabetes 2015, 7, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.B.; Wang, C.; Sørensen, K.; Bang-Berthelsen, C.H.; Hansen, L.; Andersen, M.-L.M.; Hougaard, P.; Juul, A.; Zhang, C.-Y.; Pociot, F.; et al. Circulating levels of microRNA from children with newly diagnosed type 1 diabetes and healthy controls: Evidence that miR-25 associates to residual beta-cell function and glycaemic control during disease progression. Exp. Diabetes Res. 2012, 2012, 896362. [Google Scholar] [CrossRef] [Green Version]
- Satake, E.; Pezzolesi, M.G.; Md Dom, Z.I.; Smiles, A.M.; Niewczas, M.A.; Krolewski, A.S. Circulating miRNA profiles associated with hyperglycemia in patients with Type 1 Diabetes. Diabetes 2018, 67, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipova, J.; Fischer, D.-C.; Dangwal, S.; Volkmann, I.; Widera, C.; Schwarz, K.; Lorenzen, J.M.; Schreiver, C.; Jacoby, U.; Heimhalt, M.; et al. Diabetes-Associated MicroRNAs in pediatric patients with type 1 diabetes mellitus: A cross-sectional cohort study. J. Clin. Endocrinol. Metab. 2014, 99, E1661–E1665. [Google Scholar] [CrossRef] [Green Version]
- Lanza, I.R.; Zhang, S.; Ward, L.E.; Karakelides, H.; Raftery, D.; Nair, K.S. Quantitative metabolomics by H-NMR and LC-MS/MS confirms altered metabolic pathways in diabetes. PLoS ONE 2010, 5, e10538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galderisi, A.; Pirillo, P.; Moret, V.; Stocchero, M.; Gucciardi, A.; Perilongo, G.; Moretti, C.; Monciotti, C.; Giordano, G.; Baraldi, E. Metabolomics reveals new metabolic perturbations in children with type 1 diabetes. Pediatr. Diabetes 2018, 19, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Pflueger, M.; Seppänen-Laakso, T.; Suortti, T.; Hyötyläinen, T.; Achenbach, P.; Bonifacio, E.; Orešič, M.; Ziegler, A.G. Age- and islet autoimmunity-associated differences in amino acid and lipid metabolites in children at risk for type 1 diabetes. Diabetes 2011, 60, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, V.D.N.; Lima-Neto, A.B.M.; van Tilburg, M.F.; de Oliveira Monteiro-Moreira, A.C.; Duarte Pinto Lobo, M.; Rondina, D.; Fernandes, V.O.; Montenegro, A.; Montenegro, R.M.J.; Guedes, M.I.F. Proteomic analysis to identify candidate biomarkers associated with type 1 diabetes. Diabetes Metab Syndr. Obes 2018, 11, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-Layton, D.; Guyer, P.; Delong, T.; Dang, M.; Chow, I.T.; Speake, C.; Greenbaum, C.J.; Kwok, W.W.; Baker, R.L.; Haskins, K.; et al. Hybrid insulin peptides are recognized by human T cells in the context of DRB1*04:01. Diabetes 2020, 69, 1492. [Google Scholar] [CrossRef] [PubMed]
- Colli, M.L.; Ramos-Rodríguez, M.; Nakayasu, E.S.; Alvelos, M.I.; Lopes, M.; Hill, J.L.E.; Turatsinze, J.V.; Coomans de Brachène, A.; Russell, M.A.; Raurell-Vila, H.; et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat. Commun. 2020, 11, 2584. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Qian, W.J.; Evans-Molina, C.; Mirmira, R.G.; Eizirik, D.L.; Metz, T.O. The role of proteomics in assessing beta-cell dysfunction and death in type 1 diabetes. Expert Rev. Proteom. 2019, 16, 569–582. [Google Scholar] [CrossRef]
- Suvitaival, T. Lipidomic abnormalities during the pathogenesis of type 1 diabetes: A quantitative review. Curr. Diab Rep. 2020, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, A.J.; Weir, J.M.; Jayawardana, K.; Mortensen, H.B.; Pociot, F.; Meikle, P.J. Plasma lipid species at type 1 diabetes onset predict residual beta-cell function after 6 months. Metabolomics 2018, 14, 158. [Google Scholar] [CrossRef] [Green Version]
- Oresic, M.; Gopalacharyulu, P.; Mykkänen, J.; Lietzen, N.; Mäkinen, M.; Nygren, H.; Simell, S.; Simell, V.; Hyöty, H.; Veijola, R.; et al. Cord serum lipidome in prediction of islet autoimmunity and type 1 diabetes. Diabetes 2013, 62, 3268–3274. [Google Scholar] [CrossRef] [Green Version]
- La Torre, D.; Seppänen-Laakso, T.; Larsson, H.E.; Hyötyläinen, T.; Ivarsson, S.A.; Lernmark, A.; Oresic, M.; Group, D.S. Decreased cord-blood phospholipids in young age-at-onset type 1 diabetes. Diabetes 2013, 62, 3951–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Näntö-Salonen, K.; Seppänen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Ahonen, L.; Dyrlund, T.S.; Kemppainen, E.; Siljander, H.; Hyöty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyötyläinen, T.; et al. Dynamics of plasma lipidome in progression to islet autoimmunity and type 1 diabetes-Type 1 Diabetes Prediction and Prevention Study (DIPP). Sci. Rep. 2018, 8, 10635. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Ahonen, L.; Dyrlund, T.S.; Siljander, H.; Hyöty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyötyläinen, T.; Knip, M.; et al. A longitudinal plasma lipidomics dataset from children who developed islet autoimmunity and type 1 diabetes. Sci. Data 2018, 5, 180250. [Google Scholar] [CrossRef]
- Li, C.X.; Wheelock, C.E.; Sköld, C.M.; Wheelock, Å.M. Integration of multi-omics datasets enables molecular classification of COPD. Eur. Respir. J. 2018, 51, 1701930. [Google Scholar] [CrossRef] [Green Version]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef] [Green Version]
- Speake, C.; Skinner, S.O.; Berel, D.; Whalen, E.; Dufort, M.J.; Young, W.C.; Odegard, J.M.; Pesenacker, A.M.; Gorus, F.K.; James, E.A.; et al. A composite immune signature parallels disease progression across T1D subjects. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chauhan, M.Z.; Arcuri, J.; Park, K.K.; Zafar, M.K.; Fatmi, R.; Hackam, A.S.; Yin, Y.; Benowitz, L.; Goldberg, J.L.; Samarah, M.; et al. Multi-omic analyses of growth cones at different developmental stages provides insight into pathways in adult neuroregeneration. iScience 2020, 23, 100836. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Syed, F.; Tersey, S.A.; Gritsenko, M.A.; Mitchell, H.D.; Chan, C.Y.; Dirice, E.; Turatsinze, J.V.; Cui, Y.; Kulkarni, R.N.; et al. Comprehensive proteomics analysis of stressed human islets identifies GDF15 as a target for type 1 diabetes intervention. Cell Metab. 2020, 31, 363–374.e366. [Google Scholar] [CrossRef]
- Stanfill, B.A.; Nakayasu, E.S.; Bramer, L.M.; Thompson, A.M.; Ansong, C.K.; Clauss, T.R.; Gritsenko, M.A.; Monroe, M.E.; Moore, R.J.; Orton, D.J.; et al. Quality control analysis in real-time (QC-ART): A tool for real-time quality control assessment of mass spectrometry-based proteomics data. Mol. Cell Proteom. 2018, 17, 1824–1836. [Google Scholar] [CrossRef] [Green Version]
- Alcazar, O.; Hernandez, L.F.; Nakayasu, E.S.; Piehowski, P.D.; Ansong, C.; Abdulreda, M.H.; Buchwald, P. Longitudinal proteomics analysis in the immediate microenvironment of islet allografts during progression of rejection. J. Proteom. 2020, 223, 103826. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Amidan, B.G.; Orton, D.J.; Lamarche, B.L.; Monroe, M.E.; Moore, R.J.; Venzin, A.M.; Smith, R.D.; Sego, L.H.; Tardiff, M.F.; Payne, S.H. Signatures for mass spectrometry data quality. J. Proteome Res. 2014, 13, 2215–2222. [Google Scholar] [CrossRef]
- Mayampurath, A.M.; Jaitly, N.; Purvine, S.O.; Monroe, M.E.; Auberry, K.J.; Adkins, J.N.; Smith, R.D. DeconMSn: A software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics 2008, 24, 1021–1023. [Google Scholar] [CrossRef]
- Petyuk, V.A.; Mayampurath, A.M.; Monroe, M.E.; Polpitiya, A.D.; Purvine, S.O.; Anderson, G.A.; Camp, D.G., 2nd; Smith, R.D. DtaRefinery, a software tool for elimination of systematic errors from parent ion mass measurements in tandem mass spectra data sets. Mol. Cell Proteom. 2010, 9, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [Green Version]
- Monroe, M.E.; Shaw, J.L.; Daly, D.S.; Adkins, J.N.; Smith, R.D. MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 2008, 32, 215–217. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Bultman, S.J.; Holley, D.; Hillhouse, C.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Willis, M.S. Non-targeted metabolomics of Brg1/Brm double-mutant cardiomyocytes reveals a novel role for SWI/SNF complexes in metabolic homeostasis. Metabolomics 2015, 11, 1287–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [Green Version]
- Styczynski, M.P.; Moxley, J.F.; Tong, L.V.; Walther, J.L.; Jensen, K.L.; Stephanopoulos, G.N. Systematic identification of conserved metabolites in GC/MS data for metabolomics and biomarker discovery. Anal. Chem. 2007, 79, 966–973. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Savitski, M.M.; Mathieson, T.; Zinn, N.; Sweetman, G.; Doce, C.; Becher, I.; Pachl, F.; Kuster, B.; Bantscheff, M. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013, 12, 3586–3598. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Swarup, V.; Beaulieu, J.M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 2012, 32, 10383–10395. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.W.; Kim, T.K.; Ham, H.Y.; Nam, J.S.; Kim, Y.H.; Zheng, H.; Pang, B.; Min, T.K.; Jung, J.S.; Lee, S.N.; et al. Lysophosphatidylcholine increases neutrophil bactericidal activity by enhancement of azurophil granule-phagosome fusion via glycine.GlyR alpha 2/TRPM2/p38 MAPK signaling. J. Immunol. 2010, 184, 4401–4413. [Google Scholar] [CrossRef] [PubMed]
- Assini, J.M.; Mulvihill, E.E.; Sutherland, B.G.; Telford, D.E.; Sawyez, C.G.; Felder, S.L.; Chhoker, S.; Edwards, J.Y.; Gros, R.; Huff, M.W. Naringenin prevents cholesterol-induced systemic inflammation, metabolic dysregulation, and atherosclerosis in Ldlr⁻/⁻ mice. J. Lipid Res. 2013, 54, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awata, T.; Kawasaki, E.; Tanaka, S.; Ikegami, H.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Kobayashi, T.; Iizuka, H.; Uga, M.; et al. Association of type 1 diabetes with two loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J. Clin. Endocrinol. Metab. 2009, 94, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jin, Y.; Reddy, M.V.; Podolsky, R.; Liu, S.; Yang, P.; Bode, B.; Reed, J.C.; Steed, R.D.; Anderson, S.W.; et al. Genetically dependent ERBB3 expression modulates antigen presenting cell function and type 1 diabetes risk. PLoS ONE 2010, 5, e11789. [Google Scholar] [CrossRef] [PubMed]
- Törn, C.; Hadley, D.; Lee, H.S.; Hagopian, W.; Lernmark, Å.; Simell, O.; Rewers, M.; Ziegler, A.; Schatz, D.; Akolkar, B.; et al. Role of type 1 diabetes-associated SNPs on risk of autoantibody positivity in the TEDDY study. Diabetes 2015, 64, 1818–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Contreras, M.; Tamayo-Garcia, A.; Pappan, K.L.; Michelotti, G.A.; Stabler, C.L.; Ricordi, C.; Buchwald, P. A metabolomics study of the effects of inflammation, hypoxia, and high glucose on isolated human pancreatic islets. J. Proteome Res. 2017, 16, 2294–2306. [Google Scholar] [CrossRef]
- Buchwald, P.; Tamayo-Garcia, A.; Ramamoorthy, S.; Garcia-Contreras, M.; Mendez, A.J.; Ricordi, C. A comprehensive metabolomics study to assess longitudinal biochemical changes and potential early biomarkers in NOD mice that progress to diabetes. J. Proteome Res. 2017, 16, 3873–3890. [Google Scholar] [CrossRef]
- Alcazar, O.; Hernandez, L.F.; Tschiggfrie, A.; Muehlbauer, M.J.; Bain, J.R.; Buchwald, P.; Abdulreda, M.H. Feasibility of localized metabolomics in the study of pancreatic islets and diabetes. Metabolites 2019, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.W.; Goldberg, A.L. The metabolic fates of amino acids and the formation of glutamine in skeletal muscle. J. Biol. Chem. 1978, 253, 3685–3693. [Google Scholar] [CrossRef]
- Porcellati, F.; Pampanelli, S.; Rossetti, P.; Busciantella Ricci, N.; Marzotti, S.; Lucidi, P.; Santeusanio, F.; Bolli, G.B.; Fanelli, C.G. Effect of the amino acid alanine on glucagon secretion in non-diabetic and type 1 diabetic subjects during hyperinsulinaemic euglycaemia, hypoglycaemia and post-hypoglycaemic hyperglycaemia. Diabetologia 2007, 50, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.W.; Aoyama, K.; Matsumori, Y.; Liu, J.; Swanson, R.A. Pyruvate administered after severe hypoglycemia reduces neuronal death and cognitive impairment. Diabetes 2005, 54, 1452. [Google Scholar] [CrossRef] [Green Version]
- Otto, W.; Karlfried, G.; August-Wilhelm, G. Stoffwechsel der weißen Blutzellen. Z. Für Nat. B 1958, 13, 515–516. [Google Scholar] [CrossRef]
- Fukuzumi, M.; Shinomiya, H.; Shimizu, Y.; Ohishi, K.; Utsumi, S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun. 1996, 64, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef] [PubMed]
- Morze, C.v.; Allu, P.K.R.; Chang, G.Y.; Marco-Rius, I.; Milshteyn, E.; Wang, Z.J.; Ohliger, M.A.; Gleason, C.E.; Kurhanewicz, J.; Vigneron, D.B.; et al. Non-invasive detection of divergent metabolic signals in insulin deficiency vs. insulin resistance in vivo. Sci. Rep. 2018, 8, 2088. [Google Scholar] [CrossRef]
- Adachi, Y.; De Sousa-Coelho, A.L.; Harata, I.; Aoun, C.; Weimer, S.; Shi, X.; Gonzalez Herrera, K.N.; Takahashi, H.; Doherty, C.; Noguchi, Y.; et al. l-Alanine activates hepatic AMP-activated protein kinase and modulates systemic glucose metabolism. Mol. Metab. 2018, 17, 61–70. [Google Scholar] [CrossRef]
- Erener, S.; Marwaha, A.; Tan, R.; Panagiotopoulos, C.; Kieffer, T.J. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Umu, S.U.; Langseth, H.; Bucher-Johannessen, C.; Fromm, B.; Keller, A.; Meese, E.; Lauritzen, M.; Leithaug, M.; Lyle, R.; Rounge, T.B. A comprehensive profile of circulating RNAs in human serum. RNA Biol. 2018, 15, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Flowers, E.; Kanaya, A.M.; Fukuoka, Y.; Allen, I.E.; Cooper, B.; Aouizerat, B.E. Preliminary evidence supports circulating microRNAs as prognostic biomarkers for type 2 diabetes. Obes. Sci. Pr. 2017, 3, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; You, L.; Zhu, L.; Wang, X.; Zhou, Y.; Li, Y.; Wen, J.; Xia, Y.; Ji, C.; Guo, X. Change in circulating microRNA profile of obese children indicates future risk of adult diabetes. Metabolism 2018, 78, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, G.A.; Hernandez, L.F.; Paredes, D.; Betancourt, L.R.; Abdulreda, M.H. A machine learning approach to predict pancreatic islet grafts rejection versus tolerance. PLoS ONE 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Abdulreda, M.H.; Molano, R.D.; Faleo, G.; Lopez-Cabezas, M.; Shishido, A.; Ulissi, U.; Fotino, C.; Hernandez, L.F.; Tschiggfrie, A.; Aldrich, V.R.; et al. In vivo imaging of type 1 diabetes immunopathology using eye-transplanted islets in NOD mice. Diabetologia 2019, 62, 1237–1250. [Google Scholar] [CrossRef]
- Abdulreda, M.H.; Faleo, G.; Molano, R.D.; Lopez-Cabezas, M.; Molina, J.; Tan, Y.; Echeverria, O.A.; Zahr-Akrawi, E.; Rodriguez-Diaz, R.; Edlund, P.K.; et al. High-resolution, noninvasive longitudinal live imaging of immune responses. Proc. Natl. Acad. Sci. USA 2011, 108, 12863–12868. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Abdulreda, M.H.; Cruz-Guilloty, F.; Cutrufello, N.; Shishido, A.; Martinez, R.E.; Duffort, S.; Xia, X.; Echegaray-Mendez, J.; Levy, R.B.; et al. Role of T cell recruitment and chemokine-regulated intra-graft t cell motility patterns in corneal allograft rejection. Am. J. Transpl. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Smith, H.; Ueno, Y.; Patel, T. The MicroRNA let-7a modulates interleukin-6-dependent STAT-3 survival signaling in malignant human cholangiocytes. J. Biol. Chem. 2007, 282, 8256–8264. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ahmad, T.; Sharma, A.; Mabalirajan, U.; Kulshreshtha, A.; Agrawal, A.; Ghosh, B. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 2011, 128, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.; Wang, B.; McClelland, A.; Mohan, M.; Marai, M.; Beuscart, O.; Derouiche, S.; Gray, S.; Pickering, R.; Tikellis, C.; et al. Protective effect of let-7 miRNA family in regulating inflammation in diabetes-associated atherosclerosis. Diabetes 2017, 66, 2266–2277. [Google Scholar] [CrossRef] [Green Version]
- Barseem, N.; Mahasab, M.; El Gayed, E.A. Dysregulated circulating micro RNAs markers: New evidence into expression pattern in children with T1D among Egyptian population (preprint under review for BMC Endocr. Disord.). Res. Sq. (BMC Endocr. Disord.) 2020. [Google Scholar] [CrossRef]
- Xiong, Y.; Yepuri, G.; Forbiteh, M.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy 2014, 10, 2223–2238. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: Implications for atherosclerotic plaque vulnerability. J. Am. Heart Assoc. 2013, 2, e000096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, X.F.; Rajapakse, A.G.; Yepuri, G.; Xiong, Y.; Carvas, J.M.; Ruffieux, J.; Scerri, I.; Wu, Z.; Popp, K.; Li, J.; et al. Arginase II promotes macrophage inflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. J. Am. Heart Assoc. 2012, 1, e000992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Zhao, S. miRNA-16-5p inhibits the apoptosis of high glucose-induced pancreatic β cells via targeting of CXCL10: Potential biomarkers in type 1 diabetes mellitus. Endokrynol. Pol. 2020, 71, 404–410. [Google Scholar] [CrossRef]
- Hu, Q.; Che, G.; Yang, Y.; Xie, H.; Tian, J. Histone deacetylase 3 aggravates type 1 diabetes mellitus by inhibiting lymphocyte apoptosis through the the microRNA-296-5p/Bcl-xl axis. Front. Genet. 2020, 11, 536854. [Google Scholar] [CrossRef]
- Pilemann-Lyberg, S.; Hansen, T.W.; Tofte, N.; Winther, S.A.; Theilade, S.; Ahluwalia, T.S.; Rossing, P. Uric acid is an independent risk factor for decline in kidney function, cardiovascular events, and mortality in patients with type 1 diabetes. Diabetes Care 2019, 42, 1088–1094. [Google Scholar] [CrossRef]
- Kraus, V.B. Biomarkers as drug development tools: Discovery, validation, qualification and use. Nat. Rev. Rheumatol. 2018, 14, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Borchers, C.H. Mass spectrometry based biomarker discovery, verification, and validation--quality assurance and control of protein biomarker assays. Mol. Oncol. 2014, 8, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold-Change 1 | Proteomics | Transcriptomics | Metabolomics | Lipidomics | Total |
|---|---|---|---|---|---|
| All | 2292 | 328 | 75 | 41 | 2736 |
| 1.1 | 260 | 95 | 75 | 8 | 438 |
| 1.2 | 151 | 87 | 75 | 5 | 318 |
| 1.3 | 107 | 80 | 75 | 3 | 265 |
| 2.0 | 18 | 47 | 30 | 0 | 95 |
| 3.0 | 7 | 29 | 0 | 0 | 36 |
| Expression | ||||
|---|---|---|---|---|
| Feature | Fold-Change 1 | p-Value 2 | IPA-Predicted Function/Disorder | Link to T1D |
| let-7a-5p | 1.834 | 0.133 | Inflammation of organ | |
| miR-130a-3p | 1.503 | 0.745 | Chronic inflammatory disorder | |
| miR-16-5p | 2.954 | 0.151 | Maturation of monocytes, chronic inflammatory disorder, inflammation of organ | [107] |
| miR-296-5p | 1.444 | 0.696 | Inflammation of organ | [108] |
| miR-30c-5p | 1.39 | 0.326 | Chronic inflammatory disorder, inflammation of organ | |
| miR-324-3p | 2.682 | 0.468 | Inflammation of organ | [90] |
| miR-532-5p | 2.484 | 0.122 | Chronic inflammatory disorder, inflammation of organ | |
| miR-92a-3p | 21.029 | 0.146 | Induction of follicular T helper precursor cells, chronic inflammatory disorder, inflammation of organ | |
| Uric acid | 2.008 | 0.872 | Activation of macrophages, immune response of cells, induction of peripheral blood lymphocytes, inflammation of organ | [109] |
| LPC 1-18:1(11Z) | 1.549 | 0.00949 | None | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcazar, O.; Hernandez, L.F.; Nakayasu, E.S.; Nicora, C.D.; Ansong, C.; Muehlbauer, M.J.; Bain, J.R.; Myer, C.J.; Bhattacharya, S.K.; Buchwald, P.; et al. Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes. Biomolecules 2021, 11, 383. https://doi.org/10.3390/biom11030383
Alcazar O, Hernandez LF, Nakayasu ES, Nicora CD, Ansong C, Muehlbauer MJ, Bain JR, Myer CJ, Bhattacharya SK, Buchwald P, et al. Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes. Biomolecules. 2021; 11(3):383. https://doi.org/10.3390/biom11030383
Chicago/Turabian StyleAlcazar, Oscar, Luis F. Hernandez, Ernesto S. Nakayasu, Carrie D. Nicora, Charles Ansong, Michael J. Muehlbauer, James R. Bain, Ciara J. Myer, Sanjoy K. Bhattacharya, Peter Buchwald, and et al. 2021. "Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes" Biomolecules 11, no. 3: 383. https://doi.org/10.3390/biom11030383