
Genome-Wide Identification and Characterization of the CCT Gene Family in Rapeseed (Brassica napus L.)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification, Analysis, and Chromosomal Localization of the BnaCCTs
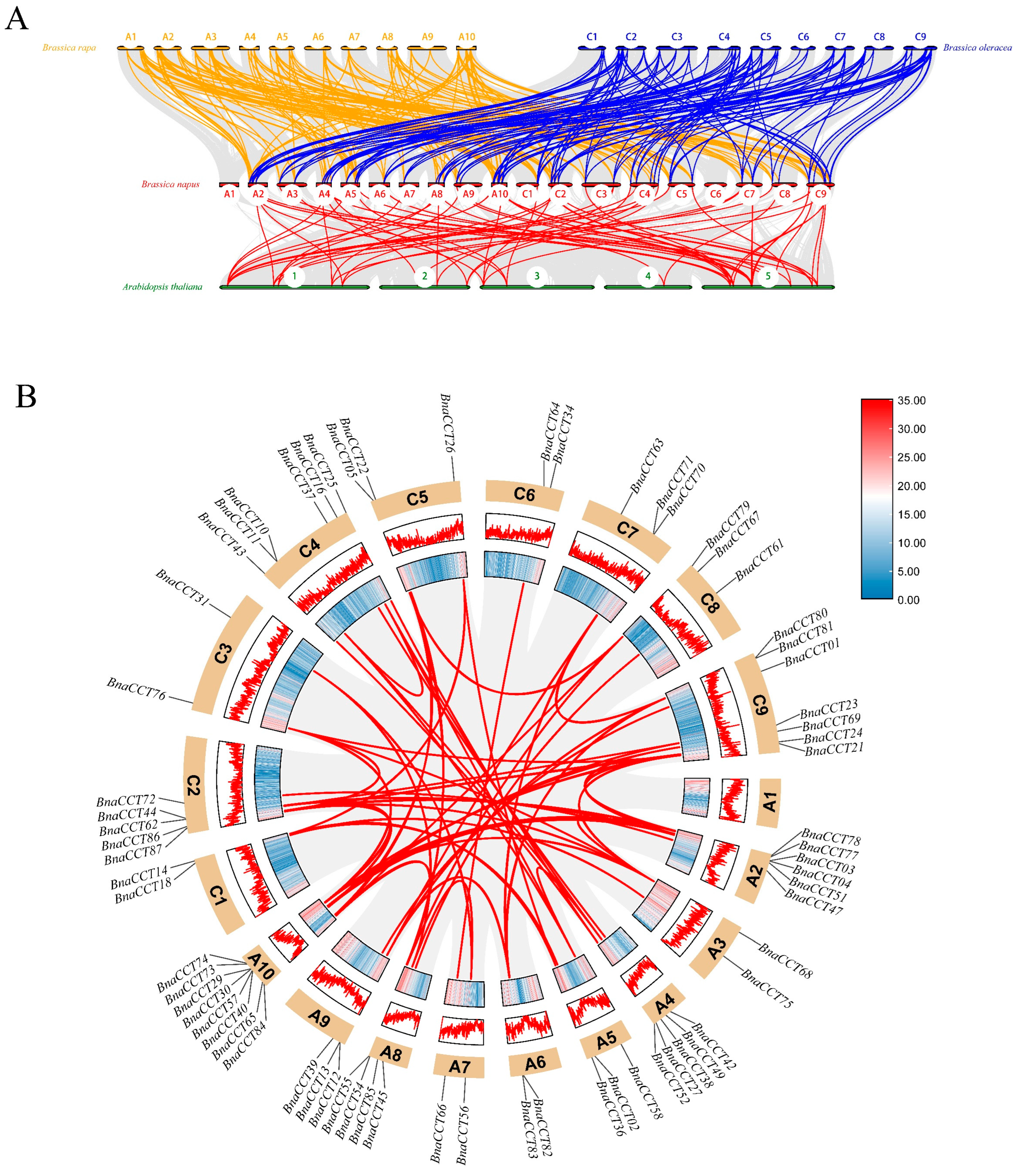
2.2. Evolution and Collinearity Analysis of the CCTs in B. napus and Other Related Species
2.3. Gene Structure, Conservative Motif Analysis, and Domain Analysis
2.4. Cis-Acting Element Analysis of the CCT Gene Family in B. napus
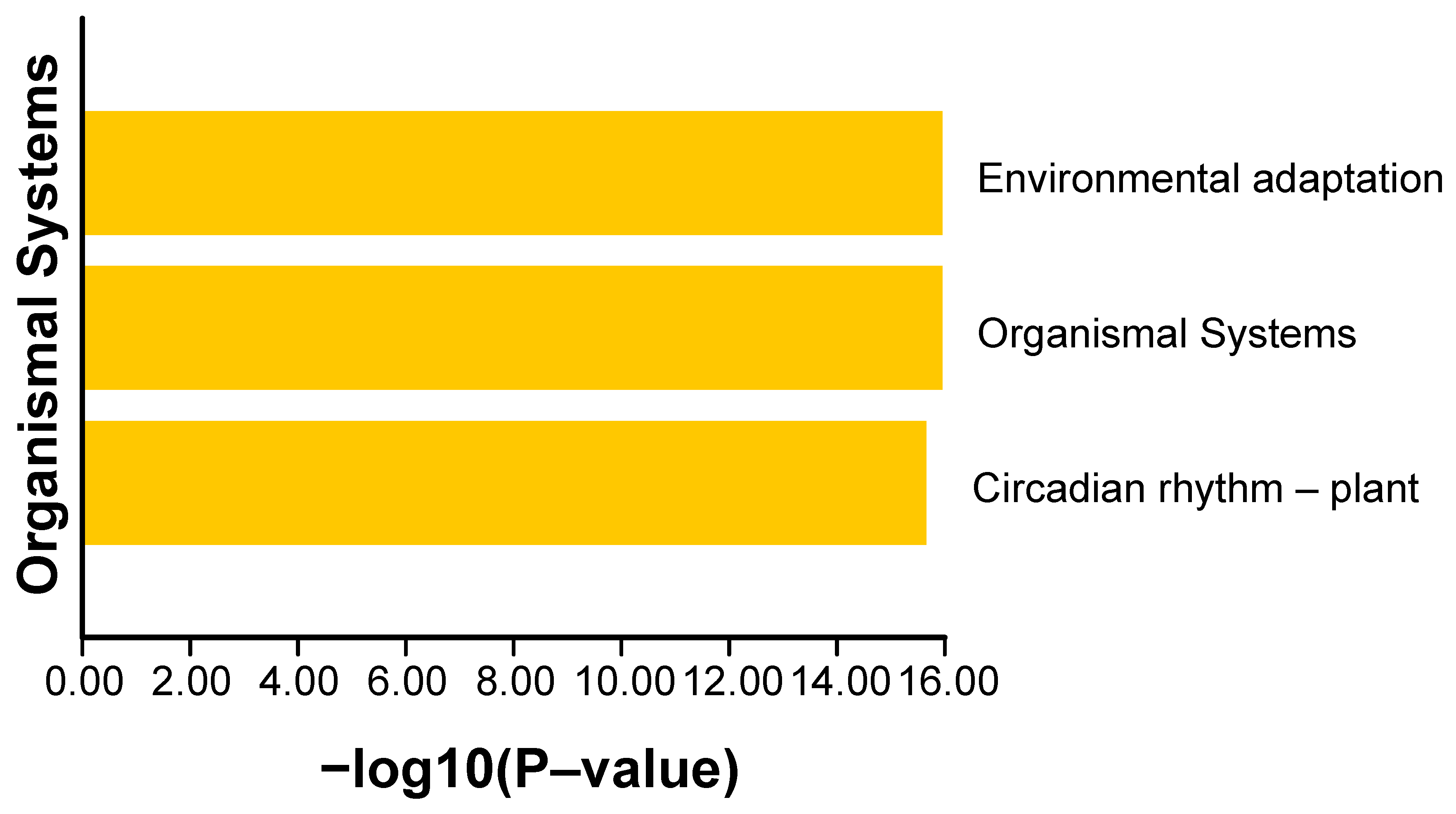
2.5. Protein Interaction Network Analysis and KEGG Enrichment Analysis of BnaCCTs
2.6. Analysis of BnaCCT Expression in Various Tissues of B. napus
2.7. Expression Analysis of the PRR Subfamily under Various Abiotic Stress Conditions and ABA Treatment
2.8. PRR Subfamily Amino Acid Sequence Alignment and Prediction of Protein Three-Dimensional Structure
2.9. qRT-PCR Analysis of the PRR Subfamily under Various Abiotic Stress Conditions and ABA Treatment
3. Discussion
4. Materials and Methods
4.1. Identification and Prediction of Physicochemical Properties of BnaCCTs
4.2. Phylogenetic Tree Analysis
4.3. Analysis of the Genetic Structure, Conserved Motifs, and Structural Domains of the BnaCCTs
4.4. Chromosome Localization and Collinearity Analysis
4.5. Cis-Acting Element Analysis and the Prediction of Subcellular Localization
4.6. Protein Interaction Network and KEGG Enrichment Analysis
4.7. Analysis of CCT Expression in B. napus
4.8. PRR Subfamily Amino Acid Sequence Alignment and Prediction of Protein Three-Dimensional Structure
4.9. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strayer, C.; Oyama, T.; Schultz, T.F.; Raman, R.; Somers, D.E.; Más, P.; Panda, S.; Kreps, J.A.; Kay, S.A. Cloning of the Arabidopsis Clock Gene TOC1, an Autoregulatory Response Regulator Homolog. Science 2000, 289, 768–771. [Google Scholar] [CrossRef]
- Robson, F.; Costa, M.M.R.; Hepworth, S.R.; Vizir, I.; Piñeiro, M.; Reeves, P.H.; Putterill, J.; Coupland, G. Functional importance of conserved domains in the flowering-time gene CONSTANS demonstrated by analysis of mutant alleles and transgenic plants. Plant J. 2022, 28, 619–631. [Google Scholar] [CrossRef]
- Li, Z.; Li, Q.P.; Dong, H.J.; He, Q.; Liang, L.; Tan, C.; Han, Z.M.; Yao, W.; Li, G.W.; Zhao, H.; et al. Three CCT domain-containing genes were identified to regulate heading date by candidate gene-based association mapping and transformation in rice. Sci. Rep. 2015, 5, 7663. [Google Scholar]
- Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Bailey, P.C.; O’Sullivan, D.M. Genome Dynamics Explain the Evolution of Flowering Time CCT Domain Gene Families in the Poaceae. PLoS ONE 2012, 7, e45307. [Google Scholar] [CrossRef]
- Yuan, X.L.; Wang, Z.S.; Jia, X.P.; Sang, L.M.; Li, J.F.; Zhang, B. Research advances on molecular mechanisms of photoperiod-regulation plant flowering and CCT gene family. J. Zhejiang Agric. Univ. 2020, 32, 1133–1140. [Google Scholar]
- Ma, L.; Yi, D.; Yang, J.; Liu, X.; Pang, Y. Genome-Wide Identification, Expression Analysis and Functional Study of CCT Gene Family in Medicago truncatula. Plants 2020, 9, 513. [Google Scholar] [CrossRef]
- Tian, S.J. Genome-Wide Identification and Analysis of Expression Patterns under Abiotic Stress of Wheat CCT Genes Family; Northwest A&F University: Yangling, China, 2020. [Google Scholar]
- Putterill, J.; Robson, F.; Lee, K.; Simon, R.; Coupland, G. The CONSTANS gene of arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 1995, 80, 847–857. [Google Scholar] [CrossRef]
- Liu, F.; Xiao, H.; Jin, Y.H.; Sun, Q.; Huang, W.; Kang, K.C.; Huang, X.Z. Cloning and expression analysis of photoperiod regulating gene CONSTANS in ephemeral plant Arabidopsis pumila. J. Shihezi Univ. Nat. Sci. 2019, 37, 190–199. [Google Scholar]
- Zhang, J.; Fan, X.W.; Hu, Y.; Zhou, X.C.; He, Q.; Liang, L.W.; Xing, Y.Z. Global analysis of CCT family knockout mutants identifies four genes involved in regulating heading date in rice. J. Integr. Plant Biol. 2021, 63, 913–923. [Google Scholar] [CrossRef]
- Wang, H.Z. New-demand oriented oilseed rape industry developing strategy. Chin. J. Oil Crop Sci. 2018, 40, 613–617. [Google Scholar]
- Zhou, E.Q. Transgenic Verification of Candidate Genes in Flowering Time of Brassica napus L.; Huazhong Agricultural University: Wuhan, China, 2021. [Google Scholar]
- Yong, W.D.; Tan, K.H.; Xu, Z.H.; Zhu, Z.Q.; Chong, K. Study on gene regulation in the determination of flowering time in higher plants. Chin. Sci. Bull. 2000, 45, 455–466. [Google Scholar] [CrossRef]
- Zhao, D.Z.; Yong, W.D.; Chong, K.; Tan, K.H. Minireview of research advances on flowering in higher plant. Chin. Bull. Bot. 1999, 16, 157–162. [Google Scholar]
- Wang, B.Q.; Wang, G.H. Research progress on early maturation of rapeseed. Crop Res. 2009, 23, 336–338. [Google Scholar]
- Fu, Y.F.; Meng, F.J. Physiological Signals of Flowering in Plants. J. China Agric. Univ. 1998, 3, 1–11. [Google Scholar]
- Liang, Y. Genome-Wide Identification of LEA and VOC Gene Families and Their Functional Mechanisms in Stress Resistance and Oil Content Formation in Brassica Napus; Huazhong University of Science and Technology: Wuhan, China, 2018. [Google Scholar]
- Liu, H.Y.; Zhou, X.C.; Li, Q.P.; Wang, L.; Xing, Y.Z. CCT domain-containing genes in cereal crops: Flowering time and beyond. Theor. Appl. Genet. 2020, 133, 1385–1396. [Google Scholar] [CrossRef]
- Mengarelli, D.A.; Zanor, M.I. Genome-wide characterization and analysis of the CCT motif family genes in soybean (Glycine max). Planta 2021, 253, 15. [Google Scholar] [CrossRef]
- Mizuno, T.; Nakamichi, N. Pseudo-Response Regulators (PRRs) or True Oscillator Components (TOCs). Plant Cell Physiol. 2005, 46, 677–685. [Google Scholar] [CrossRef]
- Ye, J.; Niu, X.; Yang, Y.; Wang, S.; Xu, Q.; Yuan, X.; Yu, H.; Wang, Y.; Wang, S.; Feng, Y.; et al. Divergent Hd1, Ghd7, and DTH7 Alleles Control Heading Date and Yield Potential of Japonica Rice in Northeast China. Front. Plant Sci. 2018, 9, 35. [Google Scholar] [CrossRef]
- Zheng, B.C. Cloning and Quantitative Expression Analyses of the Flowering Regulation Transcription Factor CONSTANS Homologous Gene in Brassica napus L.; Sichuan Agricultural University: Yaan, China, 2013. [Google Scholar]
- Guo, T. Functional Analysis of the Key Flowering Regulator BnaCO Gene in Brassica napus; Huazhong Agricultural University: Wuhan, China, 2017. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Huang, Z.N.; Duan, W.K.; Song, X.M.; Tang, J.; Wu, P.; Zhang, B.; Hou, X.L. Retention, Molecular Evolution, and Expression Divergence of the Auxin/Indole Acetic Acid and Auxin Response Factor Gene Families in Brassica rapa Shed Light on Their Evolution Patterns in Plants. Genome Biol. Evol. 2016, 8, 302–316. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Yao, M.N. Study on the Effect of BnMAPK1 on the Tolerance of Brasscia napus against Temperature Stress and the Corresponding Mechanism; Southwest University: Chongqing, China, 2020. [Google Scholar]
- Muntha, S.T.; Zhang, L.L.; Zhou, Y.F.; Zhao, X.; Hu, Z.Y.; Yang, J.H.; Zhang, M.F. Phytochrome A signal transduction 1 and CONSTANS-LIKE 13 coordinately orchestrate shoot branching and flowering in leafy Brassica juncea. Plant Biotechnol. J. 2019, 17, 1333–1343. [Google Scholar] [CrossRef]
- Chen, Y.X.; Zhou, R.J.; Hu, Q.; Wei, W.L.; Liu, J. Conservation and Divergence of the CONSTANS-Like (COL) Genes Related to Flowering and Circadian Rhythm in Brassica napus. Front. Plant Sci. 2021, 12, 760379. [Google Scholar] [CrossRef]
- Robert, L.S.; Robson, F.; Sharpe, A.; Lydiate, D.; Coupland, G. Conserved structure and function of the Arabidopsis flowering time gene CONSTANS in Brassica napus. Plant Mol. Biol. 1998, 37, 763–772. [Google Scholar] [CrossRef]
- Chao, H.Y.; Li, T.; Luo, C.Y.; Huang, H.L.; Ruan, Y.F.; Li, X.D.; Niu, Y.; Fan, Y.H.; Sun, W.; Zhang, K.; et al. BrassicaEDB: A Gene Expression Database for Brassica Crops. Int. J. Mol. Sci. 2020, 21, 5831. [Google Scholar] [CrossRef]
- Tian, M.M.; Wu, A.M.; Zhang, M.; Zhang, J.J.; Wei, H.; Yang, X.; Ma, L.; Lu, J.H.; Fu, X.K.; Wang, H.T.; et al. Genome-Wide Identification of the Early Flowering 4 (ELF4) Gene Family in Cotton and Silent GhELF4-1 and GhEFL3-6 Decreased Cotton Stress Resistance. Front. Genet. 2021, 12, 686852. [Google Scholar] [CrossRef]
- Xu, C.J.; Shan, J.M.; Liu, T.M.; Wang, Q.; Ji, Y.J.; Zhang, Y.T.; Wang, M.Y.; Xia, N.; Zhao, L. CONSTANS-LIKE 1a positively regulates salt and drought tolerance in soybean. Plant Physiol. 2023, 191, 2427–2446. [Google Scholar] [CrossRef]
- Min, J.H.; Chung, J.S.; Lee, K.H.; Kim, C.S. The CONSTANS-like 4 transcription factor, AtCOL4, positively regulates abiotic stress tolerance through an abscisic acid-dependent manner in Arabidopsis. J. Integr. Plant Biol. 2014, 57, 313–324. [Google Scholar] [CrossRef]
- Premachandran, Y. Double the action: Multimodal action of a CONSTANS-LIKE protein enhances stress tolerance in soybean. Plant Physiol. 2023, 191, 2221–2223. [Google Scholar] [CrossRef]
- Li, Y.T.; Yu, S.M.; Zhang, Q.Y.; Wang, Z.W.; Liu, M.L.; Zhang, A.; Dong, X.M.; Fan, J.J.; Zhu, Y.S.; Ruan, Y.Y.; et al. Genome-Wide Identification and Characterization of the CCT Gene Family in Foxtail Millet (Setaria italica) Response to Diurnal Rhythm and Abiotic Stress. Genes 2022, 13, 1829. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Q.; Wang, S.B.; Wei, L.L.; Huang, Y.M.; Liu, D.X.; Jia, Y.P.; Luo, C.F.; Lin, Y.C.; Liang, C.Y.; Hu, Y.; et al. BnIR: A multi-omics database with various tools for Brassica napus research and breeding. Mol. Plant. 2023, 16, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Q.; Zhu, X.Y.; Zhang, L.; Sun, X.C.; Hua, W. Effects of drought and high temperature on photosynthesis and chlorophyll fluorescence characteristics of rapeseed leaves. Chin. J. Oil Crop Sci. 2017, 39, 342–350. [Google Scholar]
- Jia, L. Effects of low temperature stress on seed germination and seedling growth of Rapeseed. Rural. Sci. Technol. 2023, 14, 86–88. [Google Scholar]
- Zheng, J.D. Effects of Soil Salt Content on Photosynthesis, Biomass Accumulation and Carbon and Nitrogen Physiology of Rapeseed; Yangzhou University: Yangzhou, China, 2023. [Google Scholar]
- Liu, E.L.; Li, Z.Q.; Luo, Z.Q.; Xu, L.L.; Jin, P.; Ji, S.; Zhou, G.H.; Wang, Z.Y.; Zhou, Z.L.; Zhang, H. Genome-Wide Identification of DUF668 Gene Family and Expression Analysis under Drought and Salt Stresses in Sweet Potato [Ipomoea batatas (L.) Lam]. Genes 2023, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.B. The Mechanism of Abscisic Acid (ABA) on Seed Oil Accumulation in Cruciferous Oilseeds; Zhejiang University: Hangzhou, China, 2019. [Google Scholar]
- Liu, H.L. Perceptions of quality improvement in oilseed Rapeseed. Crops 1992, 3, 6–7. [Google Scholar]
- Chen, H.X.; Wang, T.P.; He, X.N.; Cai, X.; Lin, R.M.; Liang, J.L.; Wu, J.; King, G.; Wang, X.W. BRAD V3.0: An upgraded Brassicaceae database. Nucleic Acids Res. 2022, 50, D1432–D1441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.Z.; Wu, D.Z.; Jiang, L.X.; Ye, L.Z. Genome-wide identification and characterization of SnRK family genes in Brassica napus. BMC Plant Biol. 2020, 20, 287. [Google Scholar] [CrossRef] [PubMed]
- Sehrish, S.; Sumbal, W.; Xie, M.L.; Zhao, C.J.; Zuo, R.; Gao, F.; Liu, S.Y. Genome-Wide Identification and Characterization of SET Domain Family Genes in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 1936. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Han, P.P.; Walk, T.C.; Yang, L.; Chen, L.Y.; Li, Y.S.; Gu, C.M.; Liao, X.; Qin, L. Genome-Wide Identification and Characterization of Ammonium Transporter (AMT) Genes in Rapeseed (Brassica napus L.). Genes 2023, 14, 658. [Google Scholar] [CrossRef]
- Heidari, P.; Hasanzadeh, S.; Faraji, S.; Ercisli, S.; Mora-Poblete, F. Genome-Wide Characterization of the Sulfate Transporter Gene Family in Oilseed Crops: Camelina sativa and Brassica napus. Plants 2023, 12, 628. [Google Scholar] [CrossRef]
- Zhang, L. The Function Verification of the Rice CCT Gene Family, mutation Mechanism Research of the New Allele OsMADS3; Huazhong Agricultural University: Hangzhou, China, 2015. [Google Scholar]
- Jin, M.L.; Liu, X.G.; Jia, W.; Liu, H.J.; Li, W.Q.; Peng, Y.; Du, Y.F.; Wang, Y.B.; Yin, Y.J.; Zhang, X.H.; et al. ZmCOL3, a CCT gene represses flowering in maize by interfering with the circadian clock and activating expression of ZmCCT. J. Integr. Plant Biol. 2018, 60, 465–480. [Google Scholar] [CrossRef]
- Yao, X.Z.; Zhang, B.H.; Zhao, D.G.; Lü, L.T. Identification and Expression Pattern Analysis of CsCCT Gene Family in Camellia sinensis L. Mol. Plant Breed. 2022, 20, 4611–4623. [Google Scholar]
- Xu, Y.; Song, S.; Wang, H.; Cao, X.; Zhao, X.; Wang, W.; Huo, L.; Li, Y.; Wassie, M.; Lu, B.; et al. Genome-wide identification of CONSTANS-LIKE (COL) family and mechanism of PpCOL8 regulating fruit senescence in sand pear (Pyrus pyrifolia)1. J. Integr. Agric. 2024, 23, 1222–1237. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Nakamichi, N.; Takao, S.; Kudo, T.; Kiba, T.; Wang, Y.; Kinoshita, T.; Sakakibara, H. Improvement of Arabidopsis Biomass and Cold, Drought and Salinity Stress Tolerance by Modified Circadian Clock-Associated PSEUDO-RESPONSE REGULATORs. Plant Cell Physiol. 2016, 57, 1085–1097. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, C.; Liang, R.Z.; Lan, M.Y.; Yu, H.; Guo, Y.H.; Chen, S.Q.; Lu, T.T.; Mo, X.; He, X.H. Genome-wide identification of the mango CONSTANS (CO) family and functional analysis of two MiCOL9 genes in transgenic Arabidopsis. Front. Plant Sci. 2022, 13, 1028987. [Google Scholar] [CrossRef]
- An, J.P.; Wang, X.F.; Zhang, X.W.; You, C.X.; Hao, Y.J. Apple B-box protein BBX37 regulates jasmonic acid mediated cold tolerance through the JAZ-BBX37-ICE1-CBF pathway and undergoes MIEL1-mediated ubiquitination and degradation. New Phytol. 2021, 229, 2707–2729. [Google Scholar] [CrossRef]
- Wang, K.; Bu, T.T.; Cheng, Q.; Dong, L.; Su, T.; Chen, Z.M.; Kong, F.J.; Gong, Z.Z.; Liu, B.H.; Li, M.N. Two homologous LHY pairs negatively control soybean drought tolerance by repressing the abscisic acid responses. New Phytol. 2021, 229, 2660–2675. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, S.J.; Min, W.K.; Cha, S.; Song, J.T.; Seo, H.S. COP1 controls salt stress tolerance by modulating sucrose content. Plant Signal. Behav. 2022, 17, e2096784. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Abe, H.; Takase, T.; Kobayashi, M.; Kiyosue, T. Overexpression of LOV KELCH PROTEIN 2 confers dehydration tolerance and is associated with enhanced expression of dehydration-inducible genes in Arabidopsis thaliana. Plant Cell Rep. 2015, 34, 843–852. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Xie, J.M.; Chen, Y.R.; Cai, G.J.; Cai, R.L.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Wang, Y.P.; Tang, H.B.; Debarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.H.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Qu, C.M.; Fu, F.Y.; Lu, K.; Zhang, K.; Wang, R.; Xu, X.F.; Wang, M.; Lu, J.X.; Wan, H.F.; Tang, Z.L.; et al. Differential accumulation of phenolic compounds and expression of related genes in black- and yellow-seeded Brassica napus. J. Exp. Bot. 2013, 64, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Bhullar, N.K. Selection of Suitable Reference Genes for qRT-PCR Gene Expression Studies in Rice. Methods Mol. Biol. 2021, 2238, 293–312. [Google Scholar]
- Swift, M.L. GraphPad Prism, Data Analysis, and Scientific Graphing. J. Chem. Inf. Modeling 1997, 37, 411–412. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Xia, J.; Jiang, R.; Wang, J.; Yuan, X.; Dong, X.; Chen, Z.; Zhao, Z.; Wu, B.; Zhan, L.; et al. Genome-Wide Identification and Characterization of the CCT Gene Family in Rapeseed (Brassica napus L.). Int. J. Mol. Sci. 2024, 25, 5301. https://doi.org/10.3390/ijms25105301
Yu L, Xia J, Jiang R, Wang J, Yuan X, Dong X, Chen Z, Zhao Z, Wu B, Zhan L, et al. Genome-Wide Identification and Characterization of the CCT Gene Family in Rapeseed (Brassica napus L.). International Journal of Molecular Sciences. 2024; 25(10):5301. https://doi.org/10.3390/ijms25105301
Chicago/Turabian StyleYu, Liyiqi, Jichun Xia, Rujiao Jiang, Jiajia Wang, Xiaolong Yuan, Xinchao Dong, Zhenjie Chen, Zizheng Zhao, Boen Wu, Lanlan Zhan, and et al. 2024. "Genome-Wide Identification and Characterization of the CCT Gene Family in Rapeseed (Brassica napus L.)" International Journal of Molecular Sciences 25, no. 10: 5301. https://doi.org/10.3390/ijms25105301