
Genotyping Hepatitis B virus by Next-Generation Sequencing: Detection of Mixed Infections and Analysis of Sequence Conservation
 ,
,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
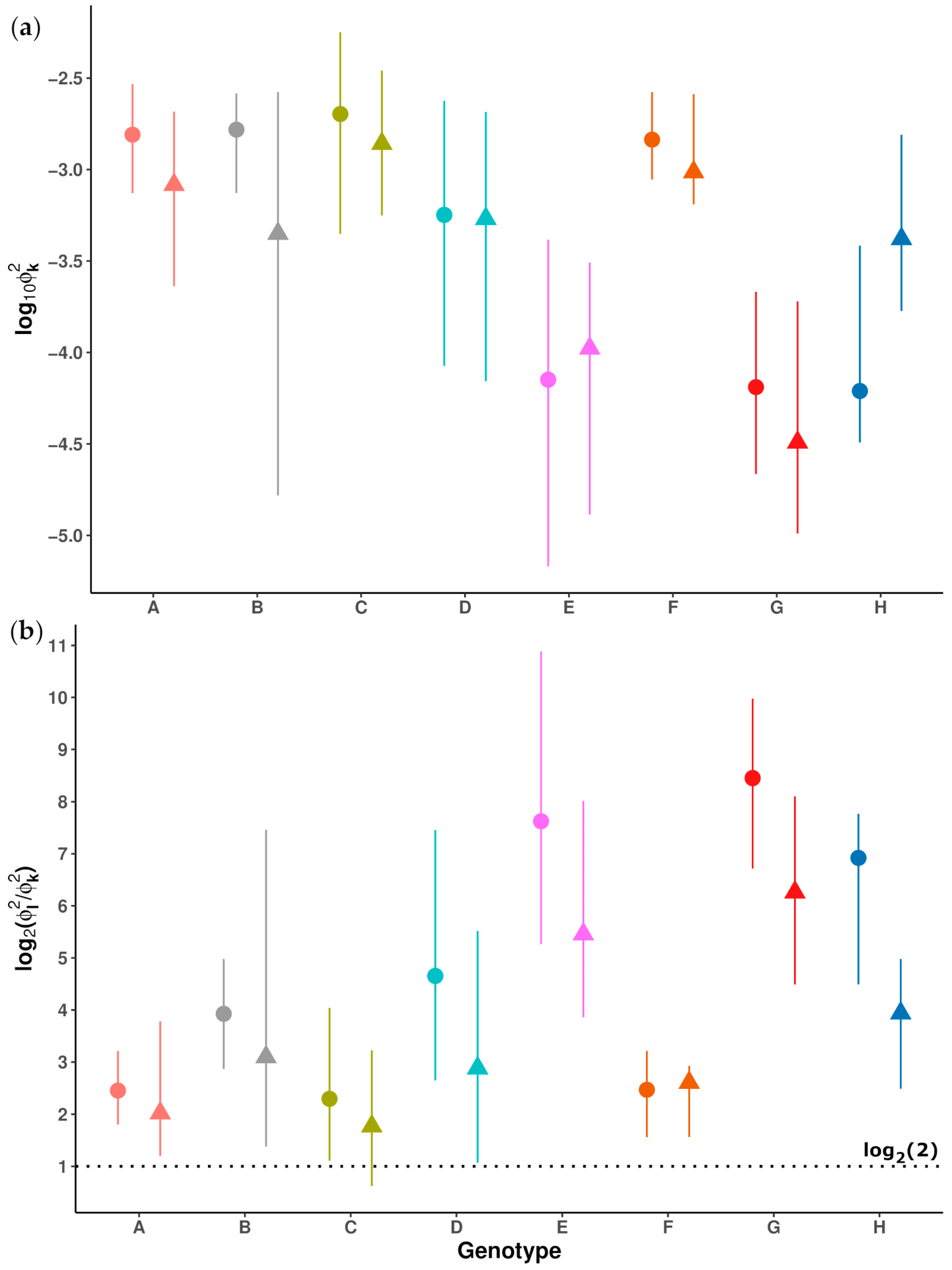
2.1. Hepatitis B Virus Genomic Regions Analyzed and Genotype Determination Using the Distance-Based Discrimination Method
2.2. Analysis of Hepatitis B Virus Genotypes
2.2.1. Next-Generation Sequencing Yield and Haplotype Genotyping
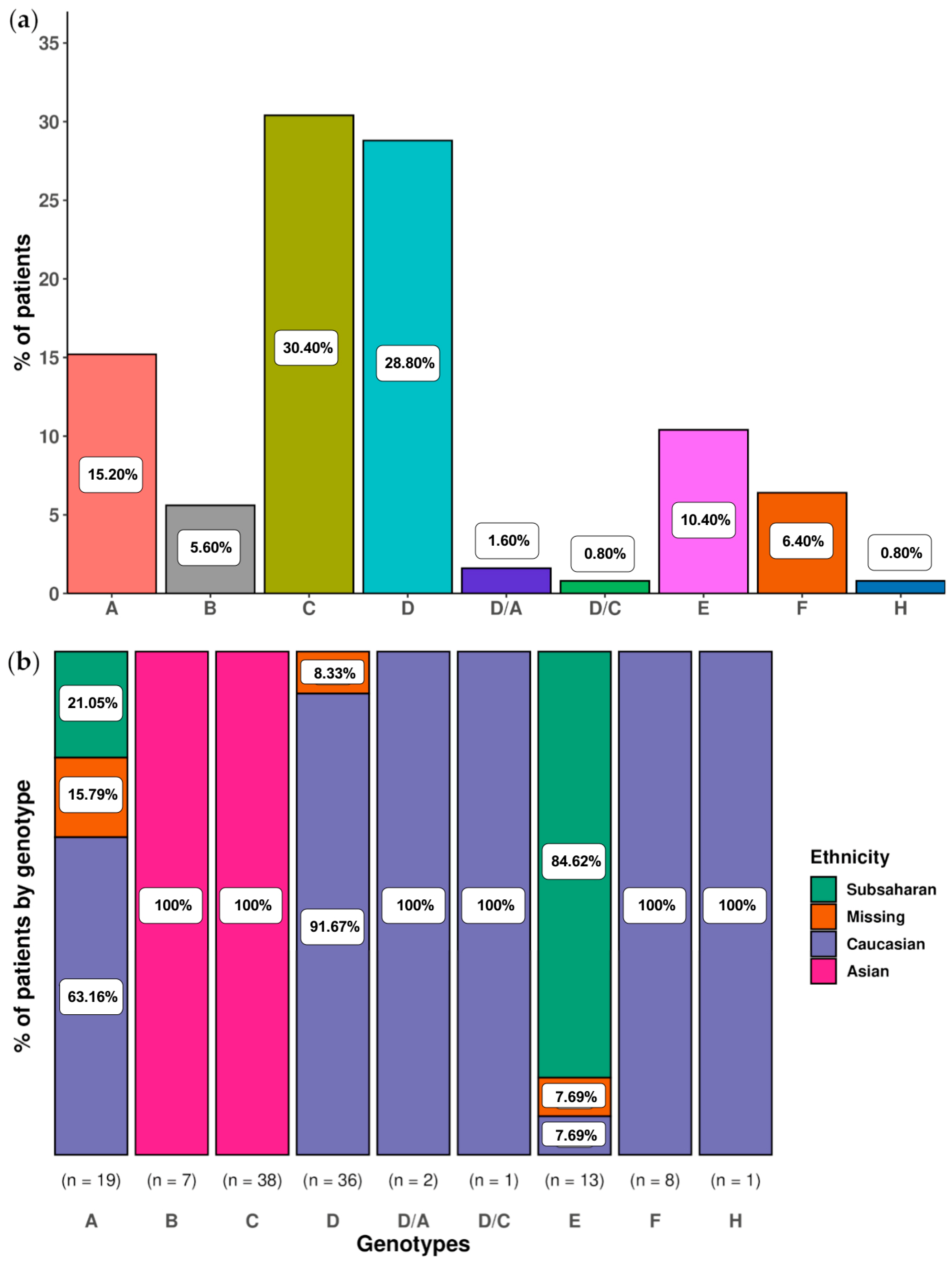
2.2.2. Analysis of Main Hepatitis B Virus Genotypes and Genotype Mixtures in Chronic Hepatitis B Patients
2.3. Study of Sequence Conservation in the preS and 5X Amplicons across Different Genotypes
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Serological and Virological Determinations
4.3. Amplification and Next-Generation Sequencing of the Hepatitis B Virus Genome
4.4. Sequencing Hepatitis B Virus Amplicons with the Sanger Method
4.5. Bionformatics Analysis of Sequencing Data
4.5.1. Processing the Next-Generation Sequencing Raw Data
- From fastq files generated by the instrument, full-length 5X and preS amplicons were reconstructed from the corresponding 300-bp paired end reads (R1 and R2) using FLASH [47], imposing a minimum of 20 overlapping bases between them and a maximum of 10% mismatches.
- Discarding reconstructed reads if 5% or more of their bases had Phred scores below Q30.
- Demultiplexing reads by matching the sequences of MID(1-n) and 5X or preS forward or reverse specific primers within windows of expected positions. A maximum of one difference in MID sequence and three differences in the specific primer sequences were accepted. Finally, primer sequences were trimmed, and a fasta file was generated for each combination of MID, specific primer, and strand, where identical reads were collapsed to haplotypes with corresponding frequencies derived from the read counts. The reverse haplotypes were reverse complemented.
- Multiple alignment of forward and reverse haplotypes using MUltiple Sequence Comparison by Log-Expectation (MUSCLE, version 3.8.31, Edgar R.C., Berkeley, CA, USA) software [48], followed by the removal of low-abundance haplotypes (with abundance <0.2% in one or both strands) and those unique to one strand. The common haplotypes were recollapsed and the corresponding coverage was taken as the sum of reads in both strands. These final haplotypes were termed consensus haplotypes and served as the basis of subsequent computations.
- Minor haplotypes <1% and/or with less than 100 reads were removed. This additional filtering step was applied on the consensus haplotypes to minimize the potential obtention of artifactual low frequency haplotypes in samples.
4.5.2. HBV Genotype Determination with the Distance-Based Discrimination Method and Selection of Reference Sequences
- is the squared distance from x to the i-th reference sequence of genotype t.
- is the squared distance between a pair of reference sequences (i, j) of genotype t. The distances between all pairs of reference sequences assigned to a specific genotype were used to compute its geometric variability, which corrects the distance between x and the genotype by the intraclass variability of the genotype.
4.5.3. Quasispecies Sequence Conservation
4.6. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 16 May 2023).
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular Carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A Global Scientific Strategy to Cure Hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Magnius, L.; Mason, W.S.; Taylor, J.; Kann, M.; Glebe, D.; Dény, P.; Sureau, C.; Norder, H. ICTV Virus Taxonomy Profile: Hepadnaviridae. J. Gen. Virol. 2020, 101, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Osiowy, C.; Giles, E.; Tanaka, Y.; Mizokami, M.; Minuk, G.Y. Molecular Evolution of Hepatitis B Virus over 25 Years. J. Virol. 2006, 80, 10307–10314. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hillt, A.M.; Boehmet, R.; Thomas, H.C.; Mcdadet, H. Viral Dynamics in Hepatitis B Virus Infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Frias, F.; Buti, M.; Tabernero, D.; Homs, M. Quasispecies Structure, Cornerstone of Hepatitis B Virus Infection: Mass Sequencing Approach. World J. Gastroenterol. 2013, 19, 6995–7023. [Google Scholar] [CrossRef] [PubMed]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular Identification of Hepatitis B Virus Genotypes/Subgenotypes: Revised Classification Hurdles and Updated Resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. Genotypes and Genetic Variability of Hepatitis B Virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-C.; Wang, L.-W.; Li, X.-J.; Liao, Y.-F.; Hu, X.-Y.; Gong, Z.-J. Relationship between HBV Genotypes and Anti-Viral Therapeutic Efficacy of Interferon-Alpha. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 166–171. [Google Scholar]
- Asim, M.; Sarma, M.P.; Kar, P. Etiological and Molecular Profile of Hepatocellular Cancer from India. Int. J. Cancer 2013, 133, 437–445. [Google Scholar] [CrossRef]
- Fletcher, G.J.; Eapen, C.E.; Abraham, P. Hepatitis B Genotyping: The Utility for the Clinicians. Indian J. Gastroenterol. 2020, 39, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Habbal, W.; Gärtner, B.C.; Monem, F. Identification of Optimal Target Gene Regions for Hepatitis B Virus Genotyping by DNA Sequencing. Intervirology 2013, 56, 325–336. [Google Scholar] [CrossRef]
- Enan, K.A.; Minosse, C.; El Hussein, A.R.M.; Selleri, M.; Giombini, E.; Capobianchi, M.R.; Elkhidir, I.M.; Mustafa, M.O.; Khair, O.M.; Hassan, D.A.; et al. Analysis of Hepatitis B Virus-Mixed Genotype Infection by Ultra Deep Pyrosequencing in Sudanese Patients, 2015–2016. Infection 2019, 47, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Gregori, J.; Homs, M.; Tabernero, D.; Gonzalez, C.; Quer, J.; Blasi, M.; Casillas, R.; Nieto, L.; Riveiro-Barciela, M.; et al. Complex Genotype Mixtures Analyzed by Deep Sequencing in Two Different Regions of Hepatitis B Virus. PLoS ONE 2015, 10, e0144816. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, X.; Liang, Z.; Shan, Y.; Huang, W.; Zhang, D.; Zen, A.; Zhou, X.; Zhao, Y.; Gong, X.; et al. Deep Sequencing Analysis of HBV Genotype Shift and Correlation with Antiviral Efficiency during Adefovir Dipivoxil Therapy. PLoS ONE 2015, 10, e0131337. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.F.; González, C.; Gregori, J.; Casillas, R.; Carioti, L.; Guerrero-Murillo, M.; Riveiro-Barciela, M.; Godoy, C.; Sopena, S.; Yll, M.; et al. Sophisticated Viral Quasispecies with a Genotype-Related Pattern of Mutations in the Hepatitis B X Gene of HBeAg-ve Chronically Infected Patients. Sci. Rep. 2021, 11, 4215. [Google Scholar] [CrossRef] [PubMed]
- González, C.; Tabernero, D.; Cortese, M.F.; Gregori, J.; Casillas, R.; Riveiro-Barciela, M.; Godoy, C.; Sopena, S.; Rando, A.; Yll, M.; et al. Detection of Hyper-Conserved Regions in Hepatitis B Virus X Gene Potentially Useful for Gene Therapy. World J. Gastroenterol. 2018, 24, 2095–2107. [Google Scholar] [CrossRef] [PubMed]
- Casillas, R.; Tabernero, D.; Gregori, J.; Belmonte, I.; Cortese, M.F.; González, C.; Riveiro-Barciela, M.; López, R.M.; Quer, J.; Esteban, R.; et al. Analysis of Hepatitis B Virus PreS1 Variability and Prevalence of the Rs2296651 Polymorphism in a Spanish Population. World J. Gastroenterol. 2018, 24, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Arenas, C.; Cuadras, C.M. Recent Statistical Methods Based on Distances. Contrib. Sci. 2002, 2, 183–191. [Google Scholar]
- Lau, K.C.; Osiowy, C.; Coffin, C.S. Hepatitis B Virus (HBV) Genome Detection and Genotyping in Virally Suppressed Patients Using Nested Polymerase Chain Reaction-Based Sanger Sequencing. Diagn. Microbiol. Infect. Dis. 2019, 93, 318–324. [Google Scholar] [CrossRef]
- Zhang, Q.; Liao, Y.; Chen, J.; Cai, B.; Su, Z.; Ying, B.; Lu, X.; Tao, C.; Wang, L. Epidemiology Study of HBV Genotypes and Antiviral Drug Resistance in Multi-Ethnic Regions from Western China. Sci. Rep. 2015, 5, 17413. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.T.; Balzanelli, M.G.; Distratis, P.; Lazzaro, R.; Tran, D.K.; Nguyen, K.C.D.; Bui, T.M.; Nguyen, T.T.; Pham, S.T.; Nguyen, H.S.D.; et al. Characteristics of Hepatitis B Virus Genotype and Sub-Genotype in Hepatocellular Cancer Patients in Vietnam. Diagnostics 2022, 12, 2393. [Google Scholar] [CrossRef]
- Lindström, A.; Odeberg, J.; Albert, J. Pyrosequencing for Detection of Lamivudine-Resistant Hepatitis B Virus. J. Clin. Microbiol. 2004, 42, 4788–4795. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium Taurocholate Cotransporting Polypeptide Is a Functional Receptor for Human Hepatitis B and D Virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. Natural History of Acute and Chronic Hepatitis B: The Role of HBV Genotypes and Mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255. [Google Scholar] [CrossRef]
- Michailidis, E.; Singh, K.; Kirby, K.A.; Hachiya, A.; Yoo, W.; Hong, S.P.; Kim, S.; Folk, W.R.; Sarafianos, S.G. Hepatitis B Virus Genotypic Differences Map Structurally Close to NRTI Resistance Hot Spots. Int. J. Curr. Chem. 2011, 2, 253–260. [Google Scholar] [PubMed]
- Toan, N.L.; Song, L.H.; Kremsner, P.G.; Duy, D.N.; Binh, V.Q.; Koeberlein, B.; Kaiser, S.; Kandolf, R.; Torresi, J.; Bock, C.T. Impact of the Hepatitis B Virus Genotype and Genotype Mixtures on the Course of Liver Disease in Vietnam. Hepatology 2006, 43, 1375–1384. [Google Scholar] [CrossRef]
- Garcia-Garcia, S.; Cortese, M.F.; Rodríguez-Algarra, F.; Tabernero, D.; Rando-Segura, A.; Quer, J.; Buti, M.; Rodríguez-Frías, F. Next-Generation Sequencing for the Diagnosis of Hepatitis B: Current Status and Future Prospects. Expert Rev. Mol. Diagn. 2021, 21, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Dong, P.; Zhang, K.; Deng, L.; Bach, C.; Chen, W.; Li, F.; Protzer, U.; Ding, H.; Zeng, C. Whole Genome HBV Deletion Profiles and the Accumulation of PreS Deletion Mutant during Antiviral Treatment. BMC Microbiol. 2012, 12, 307. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Verbeeck, J.; Lemey, P.; Zeller, M.; Rahman, M.; Maes, P.; Nevens, F.; Van Ranst, M. Molecular Evolutionary Analysis and Mutational Pattern of Full-Length Genomes of Hepatitis B Virus Isolated from Belgian Patients with Different Clinical Manifestations. J. Med. Virol. 2010, 82, 379–389. [Google Scholar] [CrossRef]
- Elizalde, M.M.; Tadey, L.; Mammana, L.; Quarleri, J.F.; Campos, R.H.; Flichman, D.M. Biological Characterization of Hepatitis B Virus Genotypes: Their Role in Viral Replication and Antigen Expression. Front. Microbiol. 2021, 12, 758613. [Google Scholar] [CrossRef]
- Pley, C.; Lourenço, J.; McNaughton, A.L.; Matthews, P.C. Spacer Domain in Hepatitis B Virus Polymerase: Plugging a Hole or Performing a Role? J. Virol. 2022, 96, e00051-22. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Hu, J. Hepatitis B Virus Reverse Transcriptase: Diverse Functions as Classical and Emerging Targets for Antiviral Intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef]
- Livingston, S.E.; Simonetti, J.P.; Bulkow, L.R.; Homan, C.E.; Snowball, M.M.; Cagle, H.H.; Negus, S.E.; McMahon, B.J. Clearance of Hepatitis B e Antigen in Patients with Chronic Hepatitis B and Genotypes A, B, C, D, and F. Gastroenterology 2007, 133, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Trastoy, R.; Rodríguez-Frias, F.; Muñoz-Bellido, J.L.; Melón, S.; Suárez, A.; Orduña, A.; Viciana, I.; Bernal, S.; García-Bujalance, S.; et al. GEHEP 010 Study: Prevalence and Distribution of Hepatitis B Virus Genotypes in Spain (2000–2016). J. Infect. 2020, 81, 600–606. [Google Scholar] [CrossRef]
- Bai, X.; Jia, J.A.; Fang, M.; Chen, S.; Liang, X.; Zhu, S.; Zhang, S.; Feng, J.; Sun, F.; Gao, C. Deep Sequencing of HBV Pre-S Region Reveals High Heterogeneity of HBV Genotypes and Associations of Word Pattern Frequencies with HCC. PLoS Genet. 2018, 14, e1007206. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Midgley, S. Recombination in the Genesis and Evolution of Hepatitis B Virus Genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef]
- Sauvage, V.; Boizeau, L.; Candotti, D.; Vandenbogaert, M.; Servant-Delmas, A.; Caro, V.; Laperche, S. Early MinIONTM Nanopore Single-Molecule Sequencing Technology Enables the Characterization of Hepatitis B Virus Genetic Complexity in Clinical Samples. PLoS ONE 2018, 13, e0194366. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Roberts, H.E.; Bonsall, D.; de Cesare, M.; Mokaya, J.; Lumley, S.F.; Golubchik, T.; Piazza, P.; Martin, J.B.; de Lara, C.; et al. Illumina and Nanopore Methods for Whole Genome Sequencing of Hepatitis B Virus (HBV). Sci. Rep. 2019, 9, 7081. [Google Scholar] [CrossRef]
- Martinez, M.G.; Testoni, B.; Zoulim, F. Biological Basis for Functional Cure of Chronic Hepatitis B. J. Viral Hepat. 2019, 26, 786–794. [Google Scholar] [CrossRef]
- Hui, R.W.-H.; Mak, L.Y.; Seto, W.K.; Yuen, M.F. RNA Interference as a Novel Treatment Strategy for Chronic Hepatitis B Infection. Clin. Mol. Hepatol. 2022, 28, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human Immunodeficiency Virus Type 1 Escape from RNA Interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Velkov, S.; Ott, J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Godoy, C.; Tabernero, D.; Sopena, S.; Gregori, J.; Cortese, M.F.; González, C.; Casillas, R.; Yll, M.; Rando, A.; López-Martínez, R.; et al. Characterization of Hepatitis B Virus X Gene Quasispecies Complexity in Mono-Infection and Hepatitis Delta Virus Superinfection. World J. Gastroenterol. 2019, 25, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Soria, M.E.; Gregori, J.; Chen, Q.; García-Cehic, D.; Llorens, M.; de Ávila, A.I.; Beach, N.M.; Domingo, E.; Rodríguez-Frías, F.; Buti, M.; et al. Pipeline for Specific Subtype Amplification and Drug Resistance Detection in Hepatitis C Virus. BMC Infect. Dis. 2018, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R; Version 3.6.1; R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Pagès, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. R Package; Version 2.52.0; Biostrings: Efficient Manipulation of Biological Strings; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar]
- Yll, M.; Cortese, M.F.; Guerrero-Murillo, M.; Orriols, G.; Gregori, J.; Casillas, R.; González, C.; Sopena, S.; Godoy, C.; Vila, M.; et al. Conservation and Variability of Hepatitis B Core at Different Chronic Hepatitis Stages. World J. Gastroenterol. 2020, 26, 2584–2598. [Google Scholar] [CrossRef]
- Schneider, T.D. Information Content of Individual Genetic Sequences. J. Theor. Biol. 1997, 189, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Wolfe, S.A.; Brodsky, M.H.; Zhu, L.J. MotifStack for the Analysis of Transcription Factor Binding Site Evolution. Nat. Methods 2018, 15, 8–9. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R; Version 4.3.2; R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Dinno, A. R Package; Version 1.3.5; Dunn.Test: Dunn’s Test of Multiple Comparisons Using Rank Sums; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| preS | 5X | |||||
|---|---|---|---|---|---|---|
| HBV Genotype | N Haplotypes (%) † | Median (IQR) | N Haplotypes with Gaps (%) †† | N Haplotypes (%) † | Median (IQR) | N Haplotypes with Gaps |
| A | 147 (14.27) | 7.06 (5.17–10.76) | 43 (29.25) | 226 (27.49) | 3.5 (3.13–5.37) | 0 |
| B | 44 (4.27) | 10.33 (9.52–11.09) | 2 (4.55) | 30 (3.65) | 5.5 (5.15–6.24) | 0 |
| C | 317 (30.78) | 7.41 (4.40–12.15) | 170 (53.63) | 170 (20.68) | 6.77 (5.18–10.45) | 0 |
| D | 405 (39.32) | 16.77 (10.94–30.1) | 138 (34.07) | 236 (28.71) | 6.48 (4.76–9.68) | 0 |
| E | 76 (7.38) | 41.34 (18.95–106.89) | 40 (52.63) | 77 (9.37) | 13.94 (6.6–19.68) | 0 |
| F | 22 (2.13) | 4.32 (4.01–4.7) | 0 | 40 (4.87) | 6.52 (6.01–7.32) | 0 |
| G | − | − | − | − | − | − |
| H | 19 (1.84) | 40.47 (30.88 −58.42) | 0 | 43 (5.23) | 13.81 (11.18–16.41) | 0 |
| Sample † | preS Main Genotype (% Reads of Sample) | preS Minor Genotypes (% Reads of Sample) | 5X Main Genotype (% Reads of Sample) | 5X Minor Genotypes (% Reads of Sample) |
|---|---|---|---|---|
| 6 | D (100%) | – | A (70.91%) | D (13.96%); C (8.09%); E (7.04%) |
| 7 | A (100%) | – | A (66.57%) | C (13.07%); H (11.61%); D (8.75%) |
| 9 | F (100%) | – | F (95.77%) | A (4.23%) |
| 11 | C (100%) | – | C (91.2%) | A (6.53%); D (2.27%) |
| 12 | A (100%) | – | A (92.17%) | C (7.83%) |
| 23 | C (97.17%) | F (1.59%); D (1.24%) | C (100%) | – |
| 29 | B (96.93%) | D (3.07%) | B (100%) | – |
| 33 | B (93.39%) | C (3.50%); A (1.62%); D (1.49%) | B (100%) | – |
| 63 | E (98.47%) | D (1.53%) | E (100%) | – |
| 67 | D (100%) | – | D (91.40%) | A (5.95%); C (2.66%) |
| 68 | F (100%) | – | F (92.51%) | A (7.49%) |
| 70 | D (100%) | – | D (90%) | A (10%) |
| 71 | D (100%) | – | D (90.30%) | A (9.70%) |
| 74 | A (100%) | – | A (97.26%) | D (2.74%) |
| 75 | A (100%) | – | A (95.98%) | D (2.53%); C (1.48%) |
| 78 | D (100%) | – | D (92.49%) | A (4.53%); E (1.41%); C (1.46%) |
| 82 | D (100%) | – | D (66.9%) | A (33.1%) |
| 84 | A (100%) | – | A (78.8%) | C (16.55%); D (4.65%) |
| 97 | D (98.37%) | C (1.63%) | D (100%) | – |
| 98 | D (100%) | – | D (53.64%) | F (46.36%) |
| 102 | H (94.35%) | C (5.65%) | H (100%) | – |
| 107 | B (100%) | – | B (90.41%) | C (9.59%) |
| 120 | E (79.73%) | D (20.27%) | E (100%) | – |
| 121 | F (100%) | – | F (97.98%) | C (2.02%) |
| 126 | D (97.40%) | C (2.60%) | D (100) | – |
| 131 | E (69.18%) | C (30.82%) | E (100%) | – |
| 136 | A (78.11%) | C (13.73%); D (8.16%) | A (100%) | – |
| 140 | D (100%) | – | A (51.20%) | D (30.11%); C (18.69%) |
| Primer Name | Template Specific Primer Positions | Primer Sequence (5′-3′) |
|---|---|---|
| Outer M13 PCR | ||
| preS_M13_Fw | 2822–2843 † | GTTGTAAAACGACGGCCAGTGTCACCATATTCTTGGGAACAA |
| preS_M13_Rv | 57–75 | CACAGGAAACAGCTATGACCGAACTGGAGCCACCAGCAG |
| 5X_M13_Fw | 1234–1254 | GTTGTAAAACGACGGCCAGTATGCGTGGAACCTTTGTGGCT |
| 5X_M13_Rv | 1631–1612 | CACAGGAAACAGCTATGACCATGGGCGTTCACGGTGGTCT |
| Nested MID PCR | ||
| MID(1-n)_M13_Fw | – | MID(1-n)-GTTGTAAAACGACGGCCAGT |
| MID(1-n)_M13_Rv | – | MID(1-n)-CACAGGAAACAGCTATGACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dopico, E.; Vila, M.; Tabernero, D.; Gregori, J.; Rando-Segura, A.; Pacín-Ruíz, B.; Guerrero, L.; Ubillos, I.; Martínez, M.J.; Costa, J.; et al. Genotyping Hepatitis B virus by Next-Generation Sequencing: Detection of Mixed Infections and Analysis of Sequence Conservation. Int. J. Mol. Sci. 2024, 25, 5481. https://doi.org/10.3390/ijms25105481
Dopico E, Vila M, Tabernero D, Gregori J, Rando-Segura A, Pacín-Ruíz B, Guerrero L, Ubillos I, Martínez MJ, Costa J, et al. Genotyping Hepatitis B virus by Next-Generation Sequencing: Detection of Mixed Infections and Analysis of Sequence Conservation. International Journal of Molecular Sciences. 2024; 25(10):5481. https://doi.org/10.3390/ijms25105481
Chicago/Turabian StyleDopico, Eva, Marta Vila, David Tabernero, Josep Gregori, Ariadna Rando-Segura, Beatriz Pacín-Ruíz, Laura Guerrero, Itziar Ubillos, Miguel J. Martínez, Josep Costa, and et al. 2024. "Genotyping Hepatitis B virus by Next-Generation Sequencing: Detection of Mixed Infections and Analysis of Sequence Conservation" International Journal of Molecular Sciences 25, no. 10: 5481. https://doi.org/10.3390/ijms25105481