
Identification and Analysis of lncRNA and circRNA Related to Wheat Grain Development
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Statistical Analysis of Grain-Related Traits
2.2. Identification and Prediction of lncRNA Genes and circRNAs
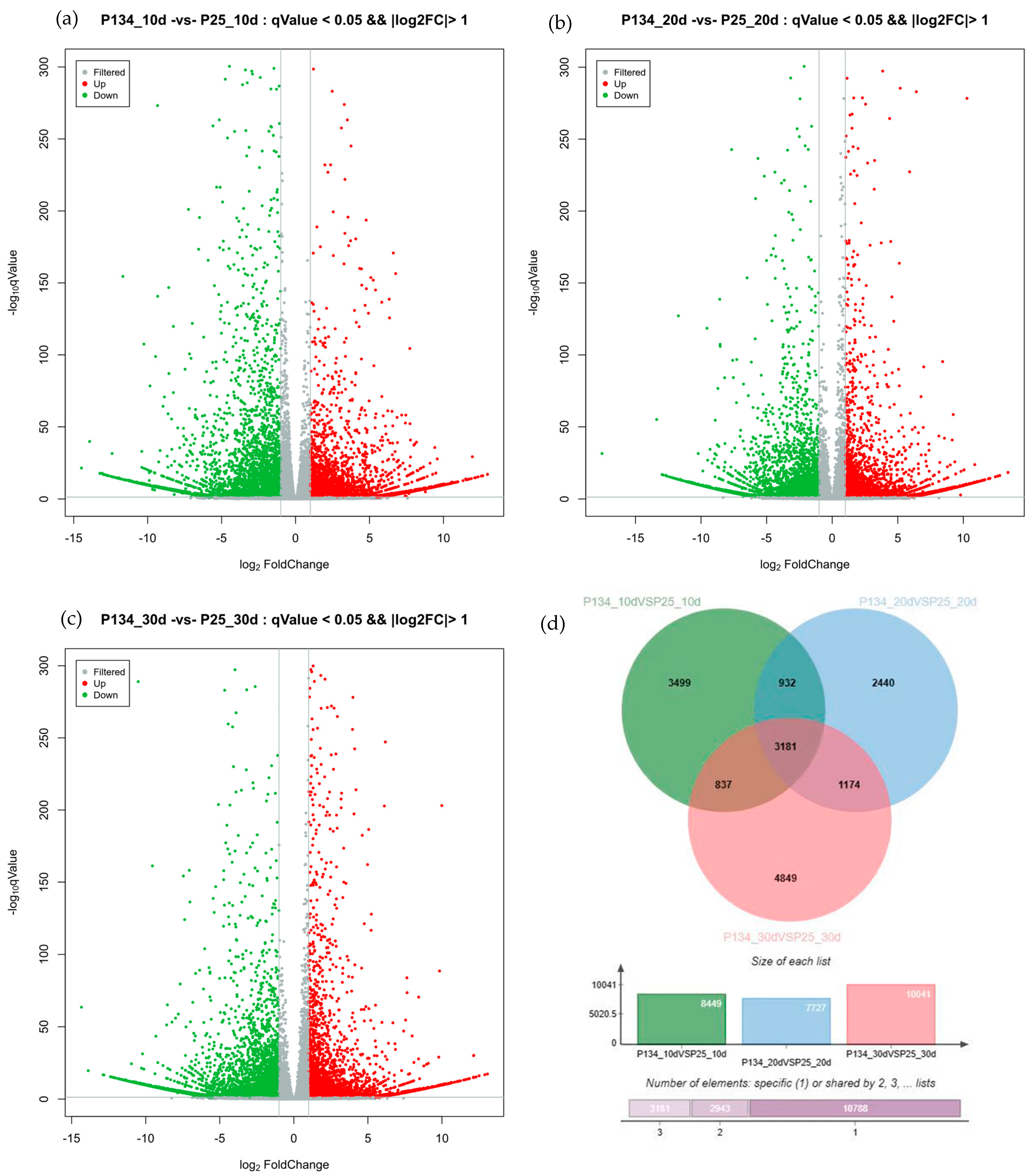
2.3. Identification of Differentially Encoded Genes and Statistical Identification of Differentially Non-Coding Genes
2.4. GO Enrichment and KEGG Enrichment Analysis of Differentially Expressed mRNA Genes
2.5. GO Enrichment and KEGG Enrichment Analysis of Differentially Expressed lncRNA Genes
2.6. GO Enrichment and KEGG Enrichment Analysis of Differentially Expressed circRNA Genes
2.7. Construction of lncRNA circRNA miRNA Interaction Network
2.8. Prediction and qRT PCR Validation of Downstream Target Genes in Interaction Modules
2.9. Analysis of the Experimental Results of Dual Luciferase
3. Materials and Methods
3.1. Plant Materials
3.2. RNA Isolation and Sequencing
3.3. Quantitative and Differential Expression Analysis of Protein-Coding Genes
3.4. Identification and Differential Expression Analysis of Non-Coding lncRNA Genes and circRNAs
3.5. Enrichment Analysis of Differential mRNA, lncRNA Neighboring Genes, and circRNA Host Genes in GO and KEGG Pathways
3.6. Prediction of lncRNA and circRNA Target Relationships and Network Construction
3.7. Quantitative Validation by qRT-PCR
3.8. Double Luciferase Experiment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, P.K.; Balyan, H.S.; Sharma, S.; Kumar, R. Genetics of yield, abiotic stress tolerance and biofortification in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2020, 133, 1569–1602. [Google Scholar] [PubMed]
- Su, Z.Q.; Hao, C.Y.; Wang, L.F.; Dong, Y.C.; Zhang, X.Y. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 122, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.J.; Zhang, H.P.; Liu, K.; Cao, J.J.; Wang, S.X.; Jiang, H.; Wu, Z.Y.; Lu, J.; Zhu, X.F.; Xia, X.C.; et al. Cloning and Characterization of TaTGW-7A Gene Associated with Grain Weight in Wheat via SLAF-seq-BSA. Front. Plant Sci. 2016, 7, 1902. [Google Scholar] [PubMed]
- Yan, X.F.; Zhao, L.; Ren, Y.; Dong, Z.D.; Cui, D.Q.; Chen, F. Genome-wide association study revealed that the TaGW8 gene was associated with kernel size in Chinese bread wheat. Sci. Rep. 2019, 9, 2702. [Google Scholar] [PubMed]
- Wang, W.; Pan, Q.L.; Tian, B.; He, F.; Chen, Y.Y.; Bai, G.H.; Akhunova, A.; Trick, H.N.; Akhunov, E. Gene editing of the wheat homologs of TONNEAU1-recruiting motif encoding gene affects grain shape and weight in wheat. Plant J. 2019, 100, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, T.; Hao, C.Y.; Wang, Y.Q.; Chen, X.H.; Zhang, X.Y. TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol. J. 2016, 14, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, J.M.; Lin, H.Q.; Chuck, G.; Faris, J.D.; Dubcovsky, J. microRNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 2017, 144, 1966–1975. [Google Scholar] [PubMed]
- Simmonds, D.H.; O’Brien, T.P. Morphological and biochemical development of the wheat endosperm. Adv. Cereal Sci. Technol. 1981, 4, 5–70. [Google Scholar]
- Jenner, C.F.; Ugalde, T.D.; Aspinall, D. The Physiology of Starch and Protein Deposition in the Endosperm of Wheat. Aust. J. Plant Physiol. 1991, 18, 211–226. [Google Scholar] [CrossRef]
- Barneix, A.J. Physiology and biochemistry of source-regulated protein accumulation in the wheat grain. J. Plant Physiol. 2007, 164, 581–590. [Google Scholar]
- Wang, S.S.; Huang, C.; Wang, Q.C.; Chao, Y.N.; Chen, F.; Sun, J.G.; Song, X. Cloning and functional of tags2 gene related to kernel size in bread wheat. Acta Agron. Sin. 2022, 08, 1926–1937. [Google Scholar]
- Cristina, D.; Ciuca, M.; Mandea, V.; Cornea, C.P. Assessment of 25 genes reported to influence thousand grain weight in winter wheat germplasm. Cereal Res. Commun. 2021, 50, 237–243. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.F.; Hao, C.Y.; Wang, K.; Wang, Y.M.; Qin, L.; An, D.G.; Li, T.; Zhang, X.Y. TaDA1, a conserved negative regulator of kernel size, has an additive effect with TaGW2 in common wheat (Triticum aestivum L.). Plant Biotechnol. J. 2020, 18, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.Y.; Yan, J.; He, Z.H.; Wu, L.; Xia, X.C. Characterization of a cell wall invertase gene TaCwi-A1 on common wheat chromosome 2A and development of functional markers. Mol. Breed. 2012, 29, 43–52. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Lyu, M.; Han, H.M.; Zhou, S.H.; Lu, Y.Q.; Liu, W.H.; Yang, X.M.; Li, X.Q.; Zhang, J.P.; Liu, X.; et al. Identification and fine mapping of alien fragments associated with enhanced grain weight from Agropyron cristatum chromosome 7P in common wheat backgrounds. Theor. Appl. Genet. 2021, 134, 3759–3772. [Google Scholar]
- Liu, G.Y.; Zhang, R.Q.; Li, S.; Ullah, R.; Yang, F.P.; Wang, Z.H.; Guo, W.L.; You, M.S.; Li, B.Y.; Xie, C.J.; et al. TaMADS29 interacts with TaNF-YB1 to synergistically regulate early grain development in bread wheat. Sci. China Life Sci. 2023, 66, 1647–1664. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.L.; Li, Y.A.; Wang, Z.Y.; Tao, S.; Sun, G.L.; Kong, X.C.; Wang, K.; Ye, X.G.; Liu, S.S.; Geng, S.F.; et al. TaIAA21 represses TaARF25-mediated expression of TaERFs required for grain size and weight development in wheat. Plant J. 2021, 108, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.R.; Liu, H.; Wang, K.K.; Liu, L.L.; Wang, S.H.; Zhao, Y.H.; Yin, J.; Li, Y.C. Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 2013, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.G.; Du, C.Y.; Gao, H.H.; Liu, S.J.; Sun, W.; Lu, H.F.; Kang, J.; Xie, Y.X.; Ma, D.Y.; Wang, C.Y. Identification of microRNAs in developing wheat grain that are potentially involved in regulating grain characteristics and the response to nitrogen levels. BMC Plant Biol. 2020, 20, 87. [Google Scholar]
- Liu, D.M.; Song, Y.; Chen, Z.X.; Yu, D.Q. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef]
- Liu, D.M.; Yu, D.Q. MicroRNA (miR396) negatively regulates expression of ceramidase-like genes in Arabidopsis. Prog. Nat. Sci. 2009, 19, 781–785. [Google Scholar] [CrossRef]
- Tang, Z.H.; Zhang, L.P.; Xu, C.G.; Yuan, S.H.; Zhang, F.T.; Zheng, Y.L.; Zhao, C.P. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol. 2012, 159, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Guo, S.Y.; Xu, Y.Y.; Li, C.H.; Zhang, Z.Y.; Zhang, D.J.; Xu, S.J.; Zhang, C.; Chong, K. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Jian, C.; Lv, J.Y.; Yan, Y.; Chi, Q.; Li, Z.J.; Wang, Q.; Zhang, J.; Liu, X.L.; Zhao, H.X. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genom. 2014, 15, 289. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Xiao, B.X.; Guo, J.M. Acting mechanisms and research methods of long noncoding RNAs. Hereditas 2013, 35, 269–280. [Google Scholar] [CrossRef]
- Zhang, Q.; Shen, B.Z.; Dai, X.K.; Mei, M.H.; Saghai Maroof, M.A.; Li, Z.B. Using bulked extremes and recessive class to map genes for photoperiod-sensitive genic male sterility in rice. Proc. Natl. Acad. Sci. USA 1994, 91, 8675–8679. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Liu, Y.H.; Zhang, F.; Feng, Y.Y.; Yu, D.L.; Lin, S.; Cao, J.S. Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Li, J.R.; Lian, B.; Gu, H.Q.; Li, Y.; Qi, Y.J. Global identification of Arabidopsis lncRNA genes reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef]
- Zhu, B.Z.; Yang, Y.F.; Li, R.; Fu, D.Q.; Wen, L.W.; Luo, Y.B.; Zhu, H.L. RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening. J. Exp. Bot. 2015, 66, 4483–4495. [Google Scholar] [CrossRef]
- Li, R.; Fu, D.Q.; Zhu, B.Z.; Luo, Y.; Zhu, H. CRISPR/Cas9-mediated mutagenesis of lncRNA1459 alters tomato fruit ripening. Plant J. 2018, 94, 513–524. [Google Scholar] [CrossRef]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 8, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Chen, L.; Liu, C.; Zhu, Q.H.; Fan, L.J. Widespread noncoding circular RNAs in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Wang, J.L.; Wei, Q.Z.; Li, B.Y.; Zhong, X.M.; Hu, T.H.; Hu, H.J.; Bao, C.J. Transcriptome-Wide Identification and Characterization of Circular RNAs in Leaves of Chinese Cabbage (Brassica rapa L. ssp., pekinensis) in Response to Calcium Deficiency-Induced Tip-burn. Sci. Rep. 2019, 9, 14544. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Wang, S.; Wu, Y.; Nie, W.H.; Yiming, A.; Huang, J.; Ahmad, I.; Zhu, B.; Chen, G.Y. Identification and Characterization of Rice Circular RNAs Responding to Xanthomonas oryzae pv, oryzae Invasion. Phytopathology 2022, 112, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA Biogenesis competes with Pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs Article The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef]
- Wang, Y.X.; Yang, M.; Wei, S.M.; Qin, F.J.; Zhao, H.J.; Suo, B. Identification of circular RNAs and their targets in leaves of Triticum aestivum L. under dehydration stress. Front. Plant Sci. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Tang, C.; Xie, Y.M.; Yu, T.; Liu, N.; Wang, Z.Q.; Woolsey, R.J.; Tang, Y.G.; Zhang, X.Z.; Qin, W.B.; Zhang, Y.; et al. m6A-dependent biogenesis of circular RNAs in male germ cells. Cell Res. 2020, 30, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Liu, Y.H.; Zhang, H.; Wang, J.Y.; Zinta, G.; Xie, S.B.; Zhu, W.M.; Nie, W.F. Genome-Wide Identification of Circular RNAs in Response to Low-Temperature Stress in Tomato Leaves. Front. Genet. 2020, 11, 591806. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Ren, Y.Z.; Lin, T.B.; Cui, D.Q. Identification and characterization of CircRNAs involved in the regulation of wheat root length. Biol. Res. 2019, 52, 19. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.K.; Wang, Z.Q.; Zhou, W.L.; Liu, Y.; Shi, H.R.; Gou, X.J.; Li, H.J.; Lin, Y.; Li, C.X.; Liu, Y.X. Genome-wide identification and characterization of circRNAs in wheat tiller. Theor. Appl. Genet. 2023, 136, 102. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; Gong, Z.Y.; Lu, H.; Chen, Y.; Deng, F.; Ren, W.J. Low Light Conditions Alter Genome-Wide Profiles of Circular RNAs in Rice Grains during Grain Filling. Plants 2022, 11, 1272. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, K.; Liu, T.T.; Shi, Y.G.; Yang, J.W.; Sun, D.Z. Identification of Wheat Circular RNAs Responsive to Drought Stress. Sci. Agric. Sicina 2022, 55, 583–4599. [Google Scholar]
- Li, N.; Liu, T.T.; Guo, F.; Yang, J.W.; Shi, Y.G.; Wang, S.G.; Sun, D.Z. Identification of long non-coding RNA-microRNA-mRNA regulatory modules and their potential roles in drought stress response in wheat (Triticum aestivum L.). Front. Plant Sci. 2022, 13, 1011064. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.W.; Guo, F.Y.; Xu, Q.H.; Cang, J. LncRNA improves cold resistance of winter wheat by interacting with miR398. Funct. Plant Biol. 2020, 47, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Adam, R.; Harold, P.; Cole, T.; Lior, P. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-The DESeq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012; Volume 165, pp. 160–174. [Google Scholar]
- Kang, Y.J.; Yang, D.C.; Kong, L. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W1. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; Eddy, S.R.; Birney, E. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998, 26, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.T.; Chen, L.F.; Du, L.P.; Su, Z.Q.; Wang, J.F.; Ye, X.G.; Qi, L.; Zhang, Z.Y. Transcript suppression of TaGW2 increased grain width and weight in bread wheat. Funct. Integr. Genom. 2014, 14, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Chi, Q.; Guo, L.J.; Ma, M.; Zhang, L.J.; Mao, H.D.; Wu, B.W.; Liu, X.L.; Ramirez-Gonzalez, R.H.; Uauy, C.; Appels, R.; et al. Global transcriptome analysis uncovers the gene co-expression regulation network and key genes involved in grain development of wheat (Triticum aestivum L.). Funct. Integr. Genom. 2019, 19, 853–866. [Google Scholar]
- Niu, K.X.; Chang, C.Y.; Zhang, M.Q.; Guo, Y.T.; Yan, Y.; Sun, H.J.; Zhang, G.L.; Li, X.M.; Gong, Y.L.; Ding, C.H.; et al. Suppressing ASPARTIC PROTEASE 1 prolongs photosynthesis and increases wheat grain weight. Nat. Plants 2023, 9, 965–977. [Google Scholar] [CrossRef]
- Luo, J.; Li, Z.; Mo, F.; Liao, Y.; Liu, Y.C.; Liu, Y. Removal of superior wheat kernels promotes filling of inferior kernels by changing carbohydrate metabolism and sink strength. Crop J. 2021, 9, 1375–1385. [Google Scholar]
- Yao, F.Q.; Li, X.H.; Wang, H.; Song, Y.N.; Li, Z.Q.; Li, X.G.; Gao, X.Q.; Zhang, X.S.; Bie, X.M. Down-expression of TaPIN1s Increases the Tiller Number and Grain Yield in Wheat. BMC Plant Biol. 2021, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Sun, F.Y.; Chen, N.; Sun, G.L.; Wang, C.Y.; Wu, D.X. MiR396 regulatory network and its expression during grain development in wheat. Protoplasma 2021, 258, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, M.; Gahlaut, V.; Nagaraju, M.; Chaudhary, S.; Kumar, A.; Tyagi, P.; Gajula, M.P.; Singh, K.P. Genome-wide identification, characterization, and expression profiling of SPX gene family in wheat. Int. J. Biol. Macromol. 2019, 140, 17–32. [Google Scholar] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wang, L.; Wang, S.; Zhang, J.; Fu, Z.; Wu, P.; Yang, A.; Wu, D.; Sun, G.; Wang, C. Identification and Analysis of lncRNA and circRNA Related to Wheat Grain Development. Int. J. Mol. Sci. 2024, 25, 5484. https://doi.org/10.3390/ijms25105484
Wang M, Wang L, Wang S, Zhang J, Fu Z, Wu P, Yang A, Wu D, Sun G, Wang C. Identification and Analysis of lncRNA and circRNA Related to Wheat Grain Development. International Journal of Molecular Sciences. 2024; 25(10):5484. https://doi.org/10.3390/ijms25105484
Chicago/Turabian StyleWang, Meng, Lu Wang, Shuanghong Wang, Junli Zhang, Zhe Fu, Panpan Wu, Anqi Yang, Dexiang Wu, Genlou Sun, and Chengyu Wang. 2024. "Identification and Analysis of lncRNA and circRNA Related to Wheat Grain Development" International Journal of Molecular Sciences 25, no. 10: 5484. https://doi.org/10.3390/ijms25105484