
A Convenient One-Pot Synthesis of a Sterically Demanding Aniline from Aryllithium Using Trimethylsilyl Azide, Conversion to β-Diketimines and Synthesis of a β-Diketiminate Magnesium Hydride Complex
 , ,
, ,  and
and
Abstract
: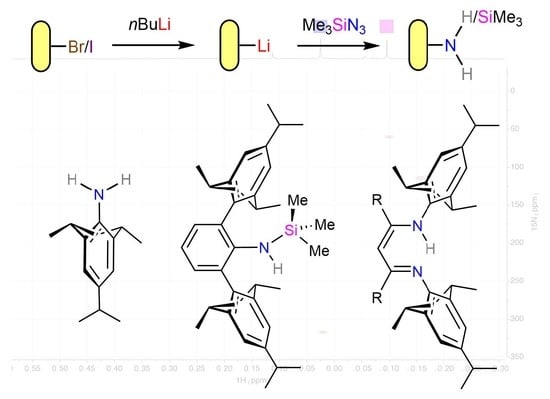
1. Introduction
2. Results and Discussion
2.1. Lithiation of TripBr
2.2. Reaction of TripLi 1 with TMSN3
2.3. Extension to a Sterically Demanding Terphenyl System
2.4. Synthesis of RTripnacnacH Compounds
2.5. Synthesis and Characterization of [{(iPrTripnacnac)MgH}2] 21
3. Conclusions
4. Materials and Methods
4.1. Experimental Details
4.2. Syntheses and Formation of Trip Compounds (1–6, 9, 10)
4.2.1. General Procedure for the Optimisation of the Lithium-Bromide Exchange
4.2.2. Synthesis of TripNH2 6
4.2.3. Data for LiN3
4.2.4. Data for TripN2N(SiMe3)2 8
4.2.5. Data for TripN(SiMe3)2 9
4.2.6. Data for TripNH3+Cl−∙1.75 H2O, 10∙1.75 H2O
4.3. Syntheses and Formation of Ter Compounds (14–17)
4.3.1. TerN(SiMe3)Li 14
4.3.2. Partial NMR Data for TerN2N(SiMe3)Li 15
4.3.3. Partial NMR Data for TerH
4.3.4. Synthesis of TerN(SiMe3)H 16
4.3.5. Deprotection of TerN(SiMe3)H 16: Synthesis of TerNH2 17
4.4. Syntheses of RTripnacnacH Compounds 18–20
4.4.1. MeTripnacnacH 18
4.4.2. EtTripnacnacH 19
4.4.3. iPrTripnacnacH 20
4.5. Synthesis of [{(iPrTripnacnac)MgH}2] 21
4.6. X-Ray Crystallographic Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsai, Y.-C. The chemistry of univalent metal β-diketiminates. Coord. Chem. Rev. 2012, 256, 722–758. [Google Scholar] [CrossRef]
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The chemistry of β-diketiminatometal complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Bulky guanidinates for the stabilization of low oxidation state metallacycles. Coord. Chem. Rev. 2010, 254, 1273–1289. [Google Scholar] [CrossRef]
- Kays, D.L. Extremely bulky amide ligands in main group chemistry. Chem. Soc. Rev. 2016, 45, 1004–1018. [Google Scholar] [CrossRef]
- Cao, C.-S.; Shi, Y.; Xu, H.; Zhao, B. Metal–metal bonded compounds with uncommon low oxidation state. Coord. Chem. Rev. 2018, 365, 122–144. [Google Scholar] [CrossRef]
- Noor, A. Coordination Chemistry of Bulky Aminopyridinates with Main Group and Transition Metals. Top. Curr. Chem. 2021, 379, 6. [Google Scholar] [CrossRef] [PubMed]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef]
- Weetman, C.; Inoue, S. The road travelled: After main-group elements as transition metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Weetman, C. Main Group Multiple Bonds for Bond Activations and Catalysis. Chem. Eur. J. 2021, 27, 1941–1954. [Google Scholar] [CrossRef]
- Forster, H.; Vögtle, F. Steric Interactions in Organic Chemistry: Spatial Requirements of Substituents. Angew. Chem. Int. Ed. Engl. 1977, 16, 429–441. [Google Scholar] [CrossRef]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef] [PubMed]
- Liptrot, D.J.; Power, P.P. London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat. Rev. Chem. 2017, 1, 0004. [Google Scholar] [CrossRef]
- Mears, K.L.; Power, P.P. Beyond Steric Crowding: Dispersion Energy Donor Effects in Large Hydrocarbon Ligands. Acc. Chem. Res. 2022, 55, 1337–1348. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Kefalidis, C.E.; Bonyhady, S.J.; Schwarze, B.; Maron, L.; Stasch, A.; Jones, C. Activation of CO by Hydrogenated Magnesium(I) Dimers: Sterically Controlled Formation of Ethenediolate and Cyclopropanetriolate Complexes. J. Am. Chem. Soc. 2015, 137, 8944–8947. [Google Scholar] [CrossRef]
- Mukherjee, D.; Okuda, J. Molecular Magnesium Hydrides. Angew. Chem. Int. Ed. 2018, 57, 1458–1473. [Google Scholar] [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable Magnesium(I) Compounds with Mg-Mg Bonds. Science 2007, 318, 1754–1757. [Google Scholar] [CrossRef]
- Rösch, B.; Gentner, T.X.; Eyselein, J.; Langer, J.; Elsen, H.; Harder, S. Strongly reducing magnesium(0) complexes. Nature 2021, 592, 717–721. [Google Scholar] [CrossRef]
- Jones, C. Dimeric magnesium(I) β-diketiminates: A new class of quasi-universal reducing agent. Nat. Rev. Chem. 2017, 1, 0059. [Google Scholar] [CrossRef]
- Queen, J.D.; Lehmann, A.; Fettinger, J.C.; Tuononen, H.M.; Power, P.P. The Monomeric Alanediyl: AlAriPr8 (AriPr8 = C6H-2,6-(C6H2-2,4,6Pri3)2-3,5-Pri2): An Organoaluminum(I) Compound with a One-Coordinate Aluminum Atom. J. Am. Chem. Soc. 2020, 142, 20554–20559. [Google Scholar] [CrossRef]
- Rit, A.; Campos, J.; Niu, H.; Aldridge, S. A stable heavier group 14 analogue of vinylidene. Nat. Chem. 2016, 8, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ruiz, D.A.; Munz, D.; Bertrand, G. A Singlet Phosphinidene Stable at Room Temperature. Chem 2016, 1, 147–153. [Google Scholar] [CrossRef]
- Wu, M.; Li, H.; Chen, W.; Wang, D.; He, Y.; Xu, L.; Ye, S.; Tan, G. A triplet stibinidene. Chem 2023, 9, 1–12. [Google Scholar] [CrossRef]
- Pang, Y.; Nöthling, N.; Leutzsch, M.; Kang, L.; Bill, E.; van Gastel, M.; Reijerse, E.; Goddard, R.; Wagner, L.; SantaLucia, D.; et al. Synthesis and isolation of a triplet bismuthinidene with a quenched magnetic response. Science 2023, 380, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Pratley, C.; Fenner, S.; Murphy, J.A. Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 2022, 122, 8181–8260. [Google Scholar] [CrossRef]
- Romanazzi, G.; Petrelli, V.; Fiore, A.M.; Mastrorilli, P.; Dell’Anna, M.M. Metal-based Heterogeneous Catalysts for One-Pot Synthesis of Secondary Anilines from Nitroarenes and Aldehydes. Molecules 2021, 26, 1120. [Google Scholar] [CrossRef]
- Saha, B.; De, S.; Dutta, S. Recent Advancements of Replacing Existing Aniline Production Process with Environmentally Friendly One-Pot Process: An Overview. Crit. Rev. Environ. Sci. Techn. 2013, 43, 84–120. [Google Scholar] [CrossRef]
- Kienle, M.; Dubbaka, S.R.; Brade, K.; Knochel, P. Modern Amination Reactions. Eur. J. Org. Chem. 2007, 2007, 4166–4176. [Google Scholar] [CrossRef]
- Cenini, S.; Gallo, E.; Caselli, A.; Ragaini, F.; Fantauzzi, S.; Piangiolino, C. Coordination chemistry of organic azides and amination reactions catalyzed by transition metal complexes. Coord. Chem. Rev. 2006, 250, 1234–1253. [Google Scholar] [CrossRef]
- Intrieri, D.; Zardi, P.; Caselli, A.; Gallo, E. Organic azides: “Energetic reagents” for the intermolecular amination of C–H bonds. Chem. Commun. 2014, 50, 11440–11453. [Google Scholar] [CrossRef]
- Corpet, M.; Gosmini, C. Recent Advances in Electrophilic Amination Reactions Electrophilic Amination Reactions. Synthesis 2014, 46, 2258–2271. [Google Scholar] [CrossRef]
- Starkov, P.; Jamison, T.F.; Marek, I. Electrophilic Amination: The Case of Nitrenoids. Chem. Eur. J. 2015, 21, 5278–5300. [Google Scholar] [CrossRef]
- Zhou, Z.; Kürti, L. Electrophilic Amination: An Update. Synlett 2019, 30, 1525–1535. [Google Scholar] [CrossRef]
- O’Neil, L.G.; Bower, J.F. Electrophilic Aminating Agents in Total Synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar] [CrossRef] [PubMed]
- Meiries, S.; Le Duc, G.; Chartoire, A.; Collado, A.; Speck, K.; Arachchige, K.S.A.; Slawin, A.M.Z.; Nolan, S.P. Large yet Flexible N-Heterocyclic Carbene Ligands for Palladium Catalysis. Chem. Eur. J. 2013, 19, 17358–17368. [Google Scholar] [CrossRef] [PubMed]
- Savka, R.; Plenio, H. Metal Complexes of Very Bulky N,N’-Diarylimidazolylidene N-Heterocyclic Carbene (NHC) Ligands with 2,4,6-Cycloalkyl Substituents. Eur. J. Inorg. Chem. 2014, 2014, 6246–6253. [Google Scholar] [CrossRef]
- Steele, B.R.; Georgakopoulos, S.; Micha-Screttas, M.; Screttas, C.G. Synthesis of New Sterically Hindered Anilines. Eur. J. Org. Chem. 2007, 2007, 3091–3094. [Google Scholar] [CrossRef]
- Oleinik, I.I.; Oleinik, I.V.; Abdrakhmanov, I.B.; Ivanchev, S.S.; Tolstikov, G.A. Design of arylimine postmetallocene catalytic systems for olefin polymerization: I. Synthesis of substituted 2-cycloalkyl- and 2,6-dicycloalkylanilines. Russ. J. Gen. Chem. 2004, 74, 1423–1427. [Google Scholar] [CrossRef]
- Atwater, B.; Chandrasoma, N.; Mitchell, D.; Rodriguez, M.J.; Pompeo, M.; Froeze, R.D.J.; Organ, M.G. The Selective Cross-Coupling of Secondary Alkyl Zinc Reagents to Five-Membered-Ring Heterocycles Using Pd-PEPPSI-IHeptCI. Angew. Chem. Int. Ed. 2015, 54, 9502–9506. [Google Scholar] [CrossRef]
- Bartlett, R.A.; Dias, H.V.R.; Power, P.P. Isolation and X-ray crystal structures of the organolithium etherate complexes, [Li(Et2O)2(CPh3)] and [{Li(Et2O)(2,4,6-(CHMe2)3C6H2)}2]. J. Organomet. Chem. 1988, 341, 1–9. [Google Scholar] [CrossRef]
- Ruhlandt-Senge, K.; Ellison, J.J.; Wehmschulte, R.J.; Pauer, F.; Power, P.P. Isolation and Structural Characterization of Unsolvated Lithium Aryls. J. Am. Chem. Soc. 1993, 115, 11353–11357. [Google Scholar] [CrossRef]
- Reich, H.J. Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms. Chem. Rev. 2013, 113, 7130–7178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, W.F.; Patricia, J.J. The Mechanism Of The Lithium–Halogen Interchange Reaction: A Review Of The Literature. J. Organomet. Chem. 1988, 352, 1–46. [Google Scholar] [CrossRef]
- Applequist, D.E.; O’Brien, D.F. Equilibria in Halogen-Lithium Interconversions. J. Am. Chem. Soc. 1962, 85, 743–748. [Google Scholar] [CrossRef]
- Jedlicka, B.; Crabtree, R.H.; Siegbahn, P.E.M. Origin of Solvent Acceleration in Organolithium Metal-Halogen Exchange Reactions. Organometallics 1997, 16, 6021–6023. [Google Scholar] [CrossRef]
- Tai, O.; Hopson, R.; Williard, P.G. Aggregation and Solvation of n-Butyllithium. Org. Lett. 2017, 19, 3966–3969. [Google Scholar] [CrossRef]
- Bailey, W.F.; Luderer, M.R.; Jordan, K.P. Effect of Solvent on the Lithium-Bromine Exchange of Aryl Bromides: Reactions of n-Butyllithium and tert-Butyllithium with 1-Bromo-4-tert-butylbenzene at 0 °C. J. Org. Chem. 2006, 71, 2825–2828. [Google Scholar] [CrossRef]
- Gorecka-Kobylinska, J.; Schlosser, M. Relative Basicities of ortho-, meta-, and para-Substituted Aryllithiums. J. Org. Chem. 2009, 74, 222–229. [Google Scholar] [CrossRef]
- Shi, L.; Chu, Y.; Knochel, P.; Mayr, H. Kinetics of Bromine-Magnesium Exchange Reactions in Substituted Bromobenzenes. J. Org. Chem. 2009, 74, 2760–2764. [Google Scholar] [CrossRef]
- Merrill, R.E.; Negishi, E.-I. Tetrahydrofuran-Promoted Aryl-Alkyl Coupling Involving Organolithium Reagents. J. Org. Chem. 1974, 39, 3452–3453. [Google Scholar] [CrossRef]
- Maercker, A. Ether Cleavage with Organo-Alkali-Metal Compounds and Alkali Metals. Angew. Chem. Int. Ed. Engl. 1987, 26, 972–989. [Google Scholar] [CrossRef]
- McGarrity, J.F.; Ogle, C.A.; Brich, Z.; Loosli, H.-R. A Rapid-Injection NMR Study of the Reactivity of Butyllithium Aggregates in Tetrahydrofuran. J. Am. Chem. Soc. 1985, 107, 1810–1815. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Application of Organic Azides for the Synthesis of Nitrogen-Containing Molecules. Synlett 2012, 1, 21–44. [Google Scholar] [CrossRef]
- Xie, S.; Sundhoro, M.; Houk, K.N.; Yan, M. Electrophilic Azides for Materials Synthesis and Chemical Biology. Acc. Chem. Res. 2020, 53, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kinjo, R. Reactions of main group compounds with azides forming organic nitrogen-containing species. Chem. Soc. Rev. 2023, 52, 5563–5606. [Google Scholar] [CrossRef] [PubMed]
- Kimball, D.B.; Haley, M.M. Triazenes: A Versatile Tool in Organic Synthesis. Angew. Chem. Int. Ed. 2002, 41, 3338–3351. [Google Scholar] [CrossRef]
- Hauber, S.-O.; Lissner, F.; Deacon, G.B.; Niemeyer, M. Stabilization of Aryl—Calcium, —Strontium, and—Barium Compounds by Designed Steric and π-Bonding Encapsulation. Angew. Chem. Int. Ed. 2005, 44, 5871–5875. [Google Scholar] [CrossRef]
- McKay, A.I.; Cole, M.L. Structural diversity in a homologous series of donor free alkali metal complexes bearing a sterically demanding triazenide. Dalton Trans. 2019, 48, 2948–2952. [Google Scholar] [CrossRef]
- Wiberg, N.; Nerudu, B. Darstellung, Eigenschaften und Struktur einiger Silylazide. Chem. Ber. 1966, 99, 740–749. [Google Scholar] [CrossRef]
- Wiberg, N.; Joo, W.-C. Zur Umsetzung von Silylaziden mit Grignard-Verbindungen. J. Organomet. Chem. 1970, 22, 333–340. [Google Scholar] [CrossRef]
- Wiberg, N.; Joo, W.-C.; Olbert, P. Zum Mechanismus der Umsetzung von Silylaziden mit Grignard-Verbindungen (Zur Frage der Reaktivität von Grignard-Verbindungen). J. Organomet. Chem. 1970, 22, 341–348. [Google Scholar] [CrossRef]
- Wiberg, N.; Joo, W.-C. Zum Mechanismus der Thermolyse von Silylazid-Grignard-Addukten (Zur Frage der Reaktivität Der N-Diazoniumgruppe). J. Organomet. Chem. 1970, 22, 349–356. [Google Scholar] [CrossRef]
- Wiberg, N.; Pracht, H.J. Darstellung und Eigenschaften einiger Silyltriazene. Chem. Ber. 1972, 105, 1377–1387. [Google Scholar] [CrossRef]
- Wiberg, N.; Pracht, H.J. Zur 1.3-Wandertendenz von Silylgruppen in Silyltriazenen. Chem. Ber. 1972, 105, 1388–1391. [Google Scholar] [CrossRef]
- Wiberg, N.; Pracht, H.J. Zur cis-trans-Isomerie von Silyltriazenen. Chem. Ber. 1972, 105, 1392–1398. [Google Scholar] [CrossRef]
- Wiberg, N.; Pracht, H.J. Zur gehinderten Rotation in Silyltriazenen. Chem. Ber. 1972, 105, 1399–1402. [Google Scholar] [CrossRef]
- Nishiyama, K.; Tanaka, N. Synthesis and reactions of trimethylsilylmethyl azide. J. Chem. Soc. Chem. Commun. 1983, 1322–1323. [Google Scholar] [CrossRef]
- Sieburth, S.M.; Somers, J.J.; O’Hare, H.K. α-Alkyl-a-aminosilanes. 1. Metalation and alkylation between silicon and nitrogen. Tetrahedron 1996, 52, 5669–5682. [Google Scholar] [CrossRef]
- Kulyabin, P.S.; Izmer, V.V.; Goryunov, G.P.; Sharikov, M.I.; Kononovich, D.S.; Uborsky, D.V.; Canich, J.-A.M.; Voskoboynikov, A.Z. Multisubstituted C2-symmetric ansa-metallocenes bearing nitrogen heterocycles: Influence of substituents on catalytic properties in propylene polymerization at higher temperatures. Dalton Trans. 2021, 50, 6170–6180. [Google Scholar] [CrossRef]
- Trost, B.M.; Pearson, W.H. Azidomethyl Phenyl Sulfide. A Synthon for NH2+. J. Am. Chem. Soc. 1981, 103, 2483–2485. [Google Scholar] [CrossRef]
- Trost, B.M.; Pearson, W.H. Sulfur activation of azides toward addition of organometallics. Amination of aliphatic carbanions. J. Am. Chem. Soc. 1983, 105, 1054–1056. [Google Scholar] [CrossRef]
- Trost, B.M.; Pearson, W.H. A synthesis of the naphthalene core of streptovaricin D via A synthon of NH2+. Tetrahedron Lett. 1983, 24, 269–272. [Google Scholar] [CrossRef]
- Krasovskiy, A.; Straub, B.F.; Knochel, P. Highly efficient Reagents for Br/Mg exchange. Angew. Chem. Int. Ed. 2005, 45, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Aoyama, T.; Shioiri, T. New Methods and Reagents in Organic Synthesis. 60. A New Synthesis of Aromatic and Heteroaromatic Amines Using Diphenyl Phosphorazidate (DPPA). Chem. Pharm. Bull. 1986, 34, 1524–1530. [Google Scholar] [CrossRef]
- Hassner, A.; Munger, P.; Belinka, B.A., Jr. A novel amination of aromatic and heteroaromatic compounds. Tetrahedron Lett. 1982, 23, 699–702. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Li, G. Synthesis of aromatic amines using allyl azide. Tetrahedron Lett. 1997, 38, 5777–5778. [Google Scholar] [CrossRef]
- Twamley, B.; Hwang, C.-S.; Hardman, N.J.; Power, P.P. Sterically encumbered terphenyl substituted primary pnictanes ArEH2 and their metallated derivatives ArE(H)Li (Ar = -C6H3-2,6-Trip2; Trip = 2,4,6-triisopropylphenyl; E = N, P, As, Sb). J. Organomet. Chem. 2000, 609, 152–160. [Google Scholar] [CrossRef]
- Wright, R.J.; Steiner, J.; Beaini, S.; Power, P.P. Synthesis of the sterically encumbering terphenyl silyl and alkyl amines HN(R)ArMes2 (R = Me and SiMe3), their lithium derivatives LiN(R)ArMes2, and the tertiary amine Me2NArMes2. Inorg. Chim. Acta 2006, 359, 1939–1946. [Google Scholar] [CrossRef]
- Gavenonis, J.; Schüwer, N.; Tilley, T.D.; Boynton, J.N.; Power, P.P. 2,6-Dimesitylaniline (H2NC6H3-2,6-Mes2) and 2,6-bis(2,4,6-triisopropylphenyl)aniline (H2NC6H3-2,6-Trip2). In Inorganic Syntheses; Power, P.P., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; Volume 37, pp. 98–105. [Google Scholar]
- Grubert, L.; Jacobi, D.; Buck, K.; Abraham, W.; Mügge, C.; Krause, E. Pseudorotaxanes and Rotaxanes Incorporating Cycloheptatrienyl Stations—Synthesis and Co-Conformation. Eur. J. Org. Chem. 2001, 2001, 3921–3932. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Zheng, Y.; Li, Y.-G.; Pan, L.; Li, Y.-S.; Hu, N.-H. Fe(II) and Co(II) pyridinebisimine complexes bearing different substituents on ortho- and para-position of imines: Synthesis, characterization and behavior of ethylene polymerization. J. Organomet. Chem. 2005, 690, 1233–1239. [Google Scholar] [CrossRef]
- Sadique, A.R.; Gregory, E.A.; Brennessel, W.W.; Holland, P.L. Mechanistic Insight into N=N Cleavage by a Low-Coordinate Iron(II) Hydride Complex. J. Am. Chem. Soc. 2007, 129, 8112–8121. [Google Scholar] [CrossRef] [PubMed]
- Hering-Junghans, C.; Schulz, A.; Thomas, M.; Villinger, A. Synthesis of mono-, di-, and triaminobismuthanes and observation of C–C coupling of aromatic systems with bismuth(III) chloride. Dalton Trans. 2016, 45, 6053–6059. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hoffmann, M.; Dreuw, A.; Hasagić, E.; Hu, C.; Stein, P.M.; Witzel, S.; Shi, H.; Yang, Y.; Rudolph, M.; et al. A metal-free direct arene C−H amination. Adv. Synth. Cat. 2021, 363, 2783–2795. [Google Scholar] [CrossRef]
- Axenrod, T.; Mangiaracina, P.; Pregosin, P.S. A 13C- and 15N-NMR. Study of Some 1-Aryl-3, 3-dimethyl Triazene Derivatives. Helv. Chim. Act. 1976, 59, 1655–1660. [Google Scholar] [CrossRef]
- Ide, S.; Iwasawa, K.; Yoshino, A.; Yoshida, T.; Takahashi, K. NMR study of the lithium salts of aniline derivatives. Magn. Reson. Chem. 1987, 25, 675–679. [Google Scholar] [CrossRef]
- Merz, K.; Bieda, R. In situ Crystallization of N(SiMe3)3 and As(SiMe3)3: Trigonal planar or pyramidal coordination of the central atoms? Z. Kristallogr. 2014, 229, 635–638. [Google Scholar] [CrossRef]
- Barnett, B.R.; Mokhtarzadeh, C.C.; Figueroa, J.S.; Lummis, P.; Wang, S.; Queen, J.D. m-Terphenyl IODO and Lithium Reagents Featuring 2,6-bis-(2,6-diisopropylphenyl) Substitution Patterns and an m-Terphenyl Lithium Etherate Featuring the 2,6-bis-(2,4,6TRIISOPROPYLPHENYL) Substitution Pattern. In Inorganic Syntheses; Power, P.P., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; Volume 37, pp. 89–98. [Google Scholar]
- Moore, D.R.; Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Mechanism of the Alternating Copolymerization of Epoxides and CO2 Using β-Diiminate Zinc Catalysts: Evidence for a Bimetallic Epoxide Enchainment. J. Am. Chem. Soc. 2003, 125, 11911–11924. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Maitland, B.; Kociok-Köhn, G.; Stasch, A.; Jones, C.; Hill, M.S. Mononuclear Three-Coordinate Magnesium Complexes of a Highly Sterically Encumbered β-Diketiminate Ligand. Inorg. Chem. 2014, 53, 10543–10552. [Google Scholar] [CrossRef]
- Ma, M.-T.; Shen, X.C.; Yu, Z.J.; Yao, W.W.; Du, L.T.; Xu, L.S.B. β-Diketiminate Magnesium Complexes: Syntheses, Crystal Structure and Catalytic Hydrosilylation. Chin. J. Inorg. Chem. 2016, 32, 1857–1866. Available online: http://www.ccspublishing.org.cn/article/doi/10.11862/CJIC.2016.232 (accessed on 7 November 2023).
- Gentner, T.X.; Rösch, B.; Ballmann, G.; Langer, J.; Elsen, H.; Harder, S. Low Valent Magnesium Chemistry with a Super Bulky β-Diketiminate Ligand. Angew. Chem. Int. Ed. 2019, 58, 607–611. [Google Scholar] [CrossRef]
- Bakhoda, A.G.; Jiang, Q.; Badiei, Y.M.; Bertke, J.A.; Cundari, T.R.; Warren, T.H. Copper-Catalyzed C(sp3)-H Amidation: Sterically Driven Primary and Secondary C-H Site-Selectivity. Angew. Chem. Int. Ed. 2019, 58, 3421–3425. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, K.; Douair, I.; Jones, D.D.L.; Maron, L.; Jones, C. Sterically controlled reductive oligomerisations of CO by activated magnesium(I) compounds: Deltate vs. ethenediolate formation. Chem. Sci. 2020, 11, 3516–3522. [Google Scholar] [CrossRef] [PubMed]
- Rösch, B.; Gentner, T.X.; Eyselein, J.; Friedrich, A.; Langer, J.; Harder, S. Mg–Mg bond polarization induced by a superbulky β-diketiminate ligand. Chem. Commun. 2020, 56, 11402–11405. [Google Scholar] [CrossRef]
- Jones, D.D.L.; Watts, S.; Jones, C. Synthesis and Characterization of Super Bulky β-Diketiminato Group 1 Metal Complexes. Inorganics 2021, 9, 72. [Google Scholar] [CrossRef]
- Mindiola, D.J.; Holland, P.L.; Warren, T.H. Complexes of Bulky β-Diketiminate Ligands. In Inorganic Syntheses; Rauchfuss, T.B., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; Volume 35, pp. 1–55. [Google Scholar]
- Burnett, S.; Bourne, C.; Slawin, A.M.Z.; van Mourik, T.; Stasch, A. Umpolung of an Aliphatic Ketone to a Magnesium Ketone‐1, 2‐diide Complex with Vicinal Dianionic Charge. Angew. Chem. Int. Ed. 2022, 61, e202204472. [Google Scholar] [CrossRef]
- López, S.E.; Restrepo, J.; Salazar, J. Polyphosphoric acid trimethylsilylester: A useful reagent for organic synthesis. J. Chem. Res. 2007, 2007, 497–502. [Google Scholar] [CrossRef]
- Yamamoto, K.; Watanabe, H. Composition of “Polyphosphoric Acid Trimethylsilyl Ester (PPSE)”and Its Use as a Condensation Reagent. Chem. Lett. 1982, 11, 1225–1228. [Google Scholar] [CrossRef]
- Roy, M.M.D.; Omanña, A.A.; Wilson, A.S.S.; Hill, M.S.; Aldridge, S.; Rivard, E. Molecular Main Group Metal Hydrides. Chem. Rev. 2021, 121, 12784–12965. Available online: https://pubs.acs.org/doi/epdf/10.1021/acs.chemrev.1c00278 (accessed on 7 November 2023). [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable Adducts of a Dimeric Magnesium(I) Compound. Angew. Chem. Int. Ed. 2008, 47, 9079–9083. [Google Scholar] [CrossRef]
- Bonyhady, S.J.; Jones, C.; Nembenna, S.; Stasch, A.; Edwards, A.J.; McIntyre, G.J. β-Diketiminate-Stabilized Magnesium(I) Dimers and Magnesium(II) Hydride Complexes: Synthesis, Characterization, Adduct Formation, and Reactivity Studies. Chem. Eur. J. 2010, 16, 938–955. [Google Scholar] [CrossRef] [PubMed]
- Treitler, D.S.; Leung, S. How Dangerous Is Too Dangerous? A Perspective on Azide Chemistry. J. Org. Chem. 2022, 87, 11293–11295. [Google Scholar] [CrossRef] [PubMed]
- Conrow, R.E.; Dean, W.D. Diazidomethane Explosion. Org. Proc. Res. Dev. 2008, 12, 1285–1286. [Google Scholar] [CrossRef]
- CrysAlisPro v1.171.42.94a Rigaku Oxford Diffraction; Rigaku Corporation: Tokyo, Japan, 2023.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. 2. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Donor, Equiv. per Li a | Temperature, T | TripBr 2 | TripH 3 (c.f. TripLi 1) | TripnBu 4 |
|---|---|---|---|---|---|
| 1 | Et2O, 17:1 | r.t. | 41.5 | 58.5 | ~0 |
| 2 | THF, 0.5:1 | r.t. | 92.2 | 7.8 | ~0 |
| 3 | THF, 1:1 | r.t. | 68.0 | 32.0 | ~0 |
| 4 | THF, 2:1 | r.t. | 23.2 | 74.8 | 2.0 |
| 5 | THF, 3:1 | r.t. | 12.9 | 83.3 | 3.8 |
| 6 | THF, 4:1 | r.t. | 9.8 | 84.7 | 5.4 |
| 7 | THF, 5:1 | r.t. | 6.6 | 85.7 | 7.7 |
| 8 | THF, 5:1 | −20 °C | trace | >97 | trace |
| 9 | THF, 5:1 (+nBuLi first) b | r.t. | 36.1 | 61.8 | 2.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demidov, N.; Grebogi, M.; Bourne, C.; McKay, A.P.; Cordes, D.B.; Stasch, A. A Convenient One-Pot Synthesis of a Sterically Demanding Aniline from Aryllithium Using Trimethylsilyl Azide, Conversion to β-Diketimines and Synthesis of a β-Diketiminate Magnesium Hydride Complex. Molecules 2023, 28, 7569. https://doi.org/10.3390/molecules28227569
Demidov N, Grebogi M, Bourne C, McKay AP, Cordes DB, Stasch A. A Convenient One-Pot Synthesis of a Sterically Demanding Aniline from Aryllithium Using Trimethylsilyl Azide, Conversion to β-Diketimines and Synthesis of a β-Diketiminate Magnesium Hydride Complex. Molecules. 2023; 28(22):7569. https://doi.org/10.3390/molecules28227569
Chicago/Turabian StyleDemidov, Nikita, Mateus Grebogi, Connor Bourne, Aidan P. McKay, David B. Cordes, and Andreas Stasch. 2023. "A Convenient One-Pot Synthesis of a Sterically Demanding Aniline from Aryllithium Using Trimethylsilyl Azide, Conversion to β-Diketimines and Synthesis of a β-Diketiminate Magnesium Hydride Complex" Molecules 28, no. 22: 7569. https://doi.org/10.3390/molecules28227569