
Discrimination of Etiologically Different Cholestasis by Modeling Proteomics Datasets

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Proteomics Analysis
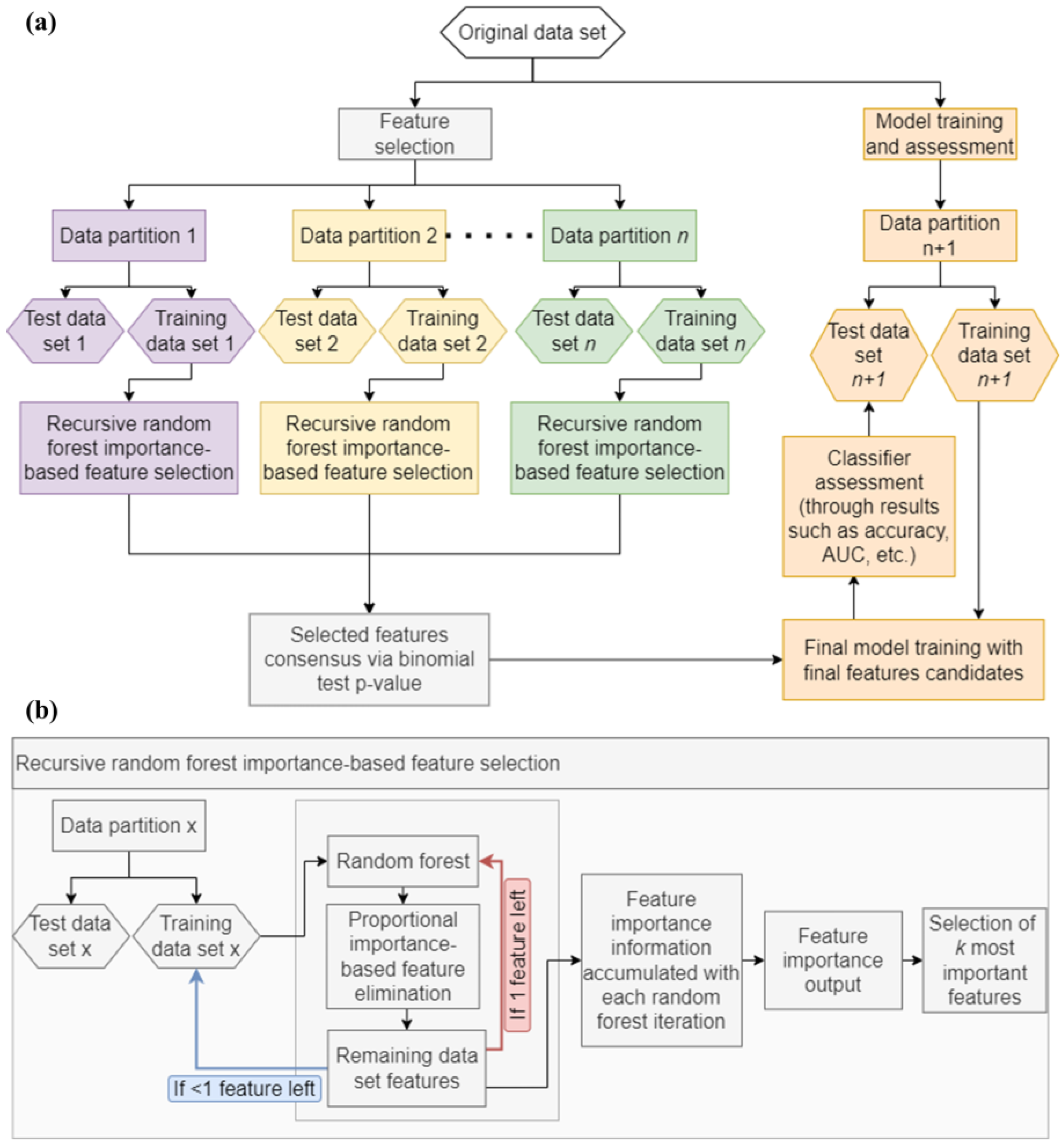
2.2. Machine Learning Analysis
3. Materials and Methods
3.1. Biological Samples
3.2. Proteomics Analysis: Sample Preparation and LC-MS/MS Conditions
3.3. Data Analysis: Proteomics Searches, Statistical and Functional Analysis
3.4. Machine Learning Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AATD | Alpha-1 antitrypsin deficiency |
| ALGS | Alagille Syndrome |
| BA | Biliary atresia |
| DDA | Data-dependent acquisition |
| DIA | Data-independent acquisition |
| KNN | K-nearest neighbors |
| LC-MS | Liquid chromatography coupled to mass spectrometry |
| NB | Naïve Bayes |
| LDA | Linear Discriminant Analysis |
| LFQ | Label-free quantification |
| PFIC | Progressive familial intrahepatic cholestasis |
| RF | Random Forest |
| SVM | Support Vector Machines |
| XGB | Extreme Gradient Boosting |
References
- Onofrio, F.Q.; Hirschfield, G.M. The Pathophysiology of Cholestasis and Its Relevance to Clinical Practice. Clin. Liver Dis. 2020, 15, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Li, M.K.; Crawford, J.M. The Pathology of Cholestasis. Semin. Liver Dis. 2004, 24, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Kerkar, N.; Todorova, L.; Kamath, B.M.; Houwen, R.H. Systematic review of progressive familial intrahepatic cholestasis. Clin. Res. Hepatol. Gastroenterol. 2018, 43, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Thompson, R.J. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Amirneni, S.; Haep, N.; Gad, A.M.; Soto-Gutierrez, A.; Squires, E.J.; Florentino, R.M. Molecular overview of progressive familial intrahepatic cholestasis. World J. Gastroenterol. 2020, 26, 7470–7484. [Google Scholar] [CrossRef]
- Fischler, B.; Lamireau, T. Cholestasis in the newborn and infant. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef]
- Mehl, A.; Bohorquez, H.; Serrano, M.-S.; Galliano, G.; Reichman, T.W. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J. Transplant. 2016, 6, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Schubert, O.T.; Röst, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289–1294. [Google Scholar] [CrossRef]
- Sandberg, A.; Branca, R.M.; Lehtiö, J.; Forshed, J. Quantitative accuracy in mass spectrometry based proteomics of complex samples: The impact of labeling and precursor interference. J. Proteom. 2014, 96, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Allmaier, G.; Apweiler, R.; Attwood, T.; Baumann, M.; Benigni, A.; Bennett, S.E.; Bischoff, R.; Bongcam-Rudloff, E.; Capasso, G.; et al. Recommendations for Biomarker Identification and Qualification in Clinical Proteomics. Sci. Transl. Med. 2010, 2, 46ps42. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.L.; Mobasheri, A.; Allaway, D.; Liddell, S.; Bacardit, J. Application of Machine Learning to Proteomics Data: Classification and Biomarker Identification in Postgenomics Biology. OMICS A J. Integr. Biol. 2013, 17, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Desaire, H.; Go, E.P.; Hua, D. Advances, obstacles, and opportunities for machine learning in proteomics. Cell Rep. Phys. Sci. 2022, 3, 101069. [Google Scholar] [CrossRef] [PubMed]
- Bauer, Y.; de Bernard, S.; Hickey, P.; Ballard, K.; Cruz, J.; Cornelisse, P.; Chadha-Boreham, H.; Distler, O.; Rosenberg, D.; Doelberg, M.; et al. Identifying early pulmonary arterial hypertension biomarkers in systemic sclerosis: Machine learning on proteomics from the DETECT cohort. Eur. Respir. J. 2020, 57, 2002591. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Carmona-Rodríguez, L.; Santos, F.M.; Ciordia, S.; Stark, L.; Hierro, L.; Pérez-Montero, P.; Vicent, D.; Corrales, F.J. Molecular basis of progressive familial intrahepatic cholestasis 3. A proteomics study. BioFactors 2024. [Google Scholar] [CrossRef] [PubMed]
- Abeel, T.; Helleputte, T.; Van de Peer, Y.; Dupont, P.; Saeys, Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 2009, 26, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Abeel, T.; Van De Peer, Y. Robust Feature Selection Using Ensemble Feature Selection Techniques. In Proceedings of the Machine Learning and Knowledge Discovery in Databases (ECML PKDD 2008), Antwerp, Belgium, 15–19 September 2008; Part II 19. pp. 313–325. [Google Scholar]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with theBorutaPackage. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Díaz-Uriarte, R.; Alvarez de Andrés, S. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kotsiantis, S.B. Decision trees: A recent overview. Artif. Intell. Rev. 2011, 39, 261–283. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Singhi, S.K.; Liu, H. Feature subset selection bias for classification learning. In Proceedings of the 23rd International Conference on Machine learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 849–856. [Google Scholar]
- Ambroise, C.; McLachlan, G.J. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc. Natl. Acad. Sci. USA 2002, 99, 6562–6566. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, N.; Ackerley, C.A.; Phillips, M.J. The structure and organization of the bile canalicular cytoskeleton with special reference to actin and actin-binding proteins. Hepatology 1995, 21, 1106–1113. [Google Scholar] [PubMed]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rodríguez, L.; Gajadhar, A.S.; Blázquez-García, I.; Guerrero, L.; Fernández-Rojo, M.A.; Uriarte, I.; Mamani-Huanca, M.; López-Gonzálvez, Á.; Ciordia, S.; Ramos, A.; et al. Mapping early serum proteome signatures of liver regeneration in living donor liver transplant cases. BioFactors 2023, 49, 912–927. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Russmann, S.; Diogo, C.; Bonfrate, L.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Mitochondria in Chronic Liver Disease. Curr. Drug Targets 2011, 12, 879–893. [Google Scholar] [CrossRef]
- Fordel, E.; Thijs, L.; Martinet, W.; Lenjou, M.; Laufs, T.; Van Bockstaele, D.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci. Lett. 2006, 410, 146–151. [Google Scholar] [CrossRef]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef]
- Wang, L.; Dong, H.; Soroka, C.J.; Wei, N.; Boyer, J.L.; Hochstrasser, M. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. J. Hepatol. 2008, 48, 1558–1569. [Google Scholar] [CrossRef]
- Capelluto, D.G. Tollip: A multitasking protein in innate immunity and protein trafficking. Microbes Infect. 2012, 14, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Paradela, A.; Corrales, F.J. Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases. Metabolites 2022, 12, 779. [Google Scholar] [CrossRef]
- Guerrero, L.; Sangro, B.; Ambao, V.; Granero, J.I.; Ramos-Fernández, A.; Paradela, A.; Corrales, F.J. Monitoring one-carbon metabolism by mass spectrometry to assess liver function and disease. J. Physiol. Biochem. 2021, 78, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Hai, N.T.T.; Thuy, L.T.T.; Shiota, A.; Kadono, C.; Daikoku, A.; Hoang, D.V.; Dat, N.Q.; Sato-Matsubara, M.; Yoshizato, K.; Kawada, N. Selective overexpression of cytoglobin in stellate cells attenuates thioacetamide-induced liver fibrosis in mice. Sci. Rep. 2018, 8, 17860. [Google Scholar] [CrossRef]
- Huang, Q.; Szklarczyk, D.; Wang, M.; Simonovic, M.; von Mering, C. PaxDb 5.0: Curated Protein Quantification Data Suggests Adaptive Proteome Changes in Yeasts. Mol. Cell. Proteom. 2023, 22, 100640. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Uklina, J. Computational Challenges in Biomarker Discovery from High-Throughput Proteomic Data. Ph.D. Thesis, ETH Zürich, Zürich, Switzerland, 2018. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (ACM, 2016), Las Vegas, NV, USA, 13–17 August 2016; Volumes 13–17, pp. 785–794. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S-PLUS; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Weihs, M.C. klaR Analyzing German Business Cycles. In Data Analysis and Decision Support; Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2005; pp. 335–343. [Google Scholar]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Meyer, D. Support Vector Machines. The Interface to libsvm in package e1071. R News 2001, 1, 23–26. [Google Scholar]
- NCAR—Research Applications Laboratory. Weather Forecast Verification Utilities, version 1.42; The Comprehensive R Archive Network (CRAN): Vienna, Austria, 2015. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot Accession | Protein Description | Gene ID | p-Value |
|---|---|---|---|
| Q9NXD2 | Myotubularin-related protein 10 | MTMR10 | <0.0001 |
| P17568 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7 | NDUFB7 | <0.0001 |
| Q15746 | Myosin light chain kinase, smooth muscle | MYLK | <0.0001 |
| Q8WWM9 | Cytoglobin | CYGB | <0.0001 |
| P48723 | Heat shock 70 kDa protein 13 | HSPA13 | <0.0001 |
| P07919 | Cytochrome b-c1 complex subunit 6, mitochondrial | UQCRH | <0.0001 |
| P01009 | Alpha-1-antitrypsin | SERPINA1 | <0.0001 |
| Q14141 | Septin-6 | SEPTIN6 | <0.0001 |
| O94874 | E3 UFM1-protein ligase 1 | UFL1 | <0.0001 |
| P22105 | Tenascin-X | TNXB | <0.0001 |
| Q9P000 | COMM domain-containing protein 9 | COMMD9 | <0.0001 |
| P03950 | Angiogenin | ANG | <0.0001 |
| Q9H223 | EH domain-containing protein 4 | EHD4 | <0.0001 |
| Q9H0E2 | Toll-interacting protein | TOLLIP | <0.0001 |
| Q9H254 | Spectrin beta chain, non-erythrocytic 4 | SPTBN4 | <0.0001 |
| Q9NR48 | Histone-lysine N-methyltransferase ASH1L | ASH1L | <0.0001 |
| P42785 | Lysosomal Pro-X carboxypeptidase | PRCP | <0.0001 |
| Q9UBR2 | Cathepsin Z | CTSZ | <0.0001 |
| P55854 | Small ubiquitin-related modifier 3 | SUMO3 | <0.0001 |
| P62829 | 60S ribosomal protein L23 | RPL23 | <0.0001 |
| Tag/ TMT Experiment | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 126 | Control | PFIC 3 | PFIC 2 | ALGS |
| 127N | PFIC 3 | Biliary atresia | ALGS | AATD |
| 127C | Biliary atresia | PFIC 3 | AATD | PFIC 1 |
| 128N | ALGS | AATD | Control | PFIC 4 |
| 128C | PFIC 3 | Control | PFIC 2 | Biliary atresia |
| 129N | Control | PFIC 2 | Biliary atresia | ALGS |
| 129C | PFIC 3 | Biliary atresia | ALGS | AATD |
| 130N | Biliary atresia | ALGS | AATD | Control |
| 130C | ALGS | AATD | Control | PFIC 4 |
| 131 | AATD | Control | PFIC 1 | Biliary atresia |
| 131C | pool | pool | pool | pool |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero, L.; Vindel-Alfageme, J.; Hierro, L.; Stark, L.; Vicent, D.; Sorzano, C.Ó.S.; Corrales, F.J. Discrimination of Etiologically Different Cholestasis by Modeling Proteomics Datasets. Int. J. Mol. Sci. 2024, 25, 3684. https://doi.org/10.3390/ijms25073684
Guerrero L, Vindel-Alfageme J, Hierro L, Stark L, Vicent D, Sorzano CÓS, Corrales FJ. Discrimination of Etiologically Different Cholestasis by Modeling Proteomics Datasets. International Journal of Molecular Sciences. 2024; 25(7):3684. https://doi.org/10.3390/ijms25073684
Chicago/Turabian StyleGuerrero, Laura, Jorge Vindel-Alfageme, Loreto Hierro, Luiz Stark, David Vicent, Carlos Óscar S. Sorzano, and Fernando J. Corrales. 2024. "Discrimination of Etiologically Different Cholestasis by Modeling Proteomics Datasets" International Journal of Molecular Sciences 25, no. 7: 3684. https://doi.org/10.3390/ijms25073684