
Using Machine Learning to Explore Shared Genetic Pathways and Possible Endophenotypes in Autism Spectrum Disorder
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
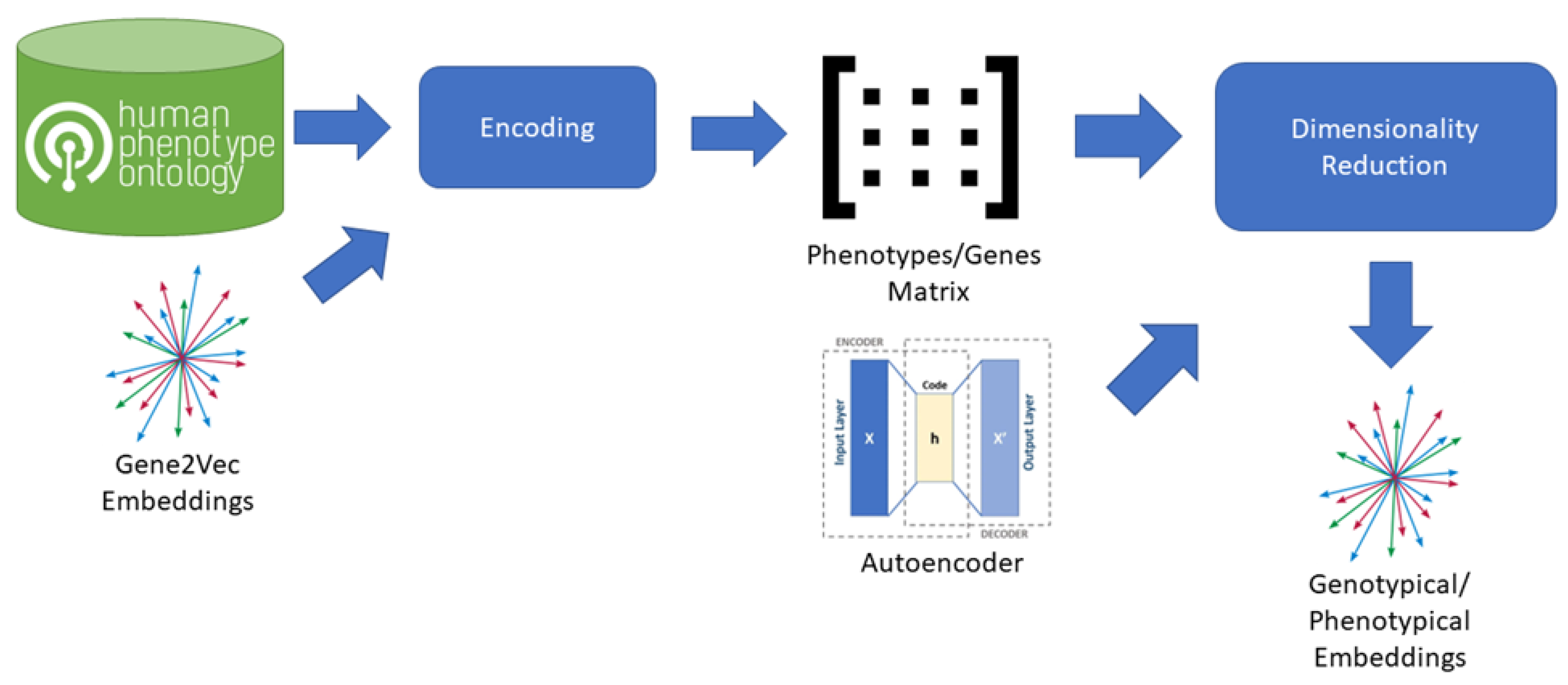
2.1. Methodological Overview
2.2. Database
2.3. Genotypical Embedding Space Creation
2.4. Phenotypical Embedding Space Creation
2.5. Dimensionality Reduction and Clustering
- min_cluster_size: the minimum number of samples a cluster should have. This parameter determines the threshold for a set of samples to be considered as noise.
- metric: the metric used to measure the distance between samples in the vectorial space. We considered ‘Euclidean’ and ‘Manhattan’.
- min_samples: the number of neighbors a sample should be close to consider it a cluster sample.
- cluster_selection_method: the way the clusters are selected in the hierarchy of clusters generated by the algorithm.
2.6. Enrichment Analysis and Additional Analyses
3. Results
3.1. Clustering Analysis
- n_neighbors = 15;
- n_components = 5;
- Metric = ‘cosine’ distance.
- Min_cluster_size: 105;
- Metric: ‘Manhattan’ distance;
- Min_samples: 10;
- Cluster_selection_method: ‘eom’ (excess of mass).
3.2. Enrichment Analysis
4. Discussion
4.1. Cluster Comparisons
4.2. Translation into Clinical Research
4.3. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Trivisano, M.; Moavero, R.; De Palma, L.; Ferretti, A.; Vigevano, F.; Curatolo, P. Autism and Epilepsy in Patients with Tuberous Sclerosis Complex. Front. Neurol. 2020, 11, 639. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhou, Y.; Wang, K. Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism. Sci. Rep. 2021, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- Giovedí, S.; Corradi, A.; Fassio, A.; Benfenati, F. Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: The case of synapsins. Front. Pediatr. 2014, 2, 94. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.; Man, H. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhong, J.; Huang, Q.; Wu, X.; Mo, X.; Lu, L.; Liang, H. Integrated Systems Analysis Explores Dysfunctional Molecular Modules and Regulatory Factors in Children with Autism Spectrum Disorder. J. Mol. Neurosci. 2021, 71, 358–368. [Google Scholar] [CrossRef]
- Li, D.; Xu, J.; Yang, M. Gene Regulation Analysis Reveals Perturbations of Autism Spectrum Disorder during Neural System Development. Genes 2021, 12, 1901. [Google Scholar] [CrossRef]
- Trivedi, P.; Pandey, M.; Rai, P.K.; Singh, P.; Srivastava, P. A meta-analysis of differentially expressed and regulatory genes with their functional enrichment analysis for brain transcriptome data in autism spectrum disorder. J. Biomol. Struct. Dyn. 2022, 1, 7. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.; Poultney, C.; Samocha, K.; Cicek, E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Trevino, A.; Müller, F.; Andersen, J.; Sundaram, L.; Kathiria, A.; Shcherbina, A.; Farh, K.; Chang, H.; Pa, A.; Kundaje, A.; et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 2021, 184, 5053–5069. [Google Scholar] [CrossRef]
- Rosina, E.; Battan, B.; Siracusano, M.; Di Criscio, L.; Hollis, F.; Pacini, L.; Curatolo, P.; Bagni, C. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry 2019, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Rizzo, D.; Hanley, J.; Coderre, E.; Prelock, P. Identifying neuroanatomical and behavioral features for autism spectrum disorder diagnosis in children using machine learning. PLoS ONE 2022, 17, e0269773. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, J.; Son, S.; Jang, Y.; Kim, B.; Lee, H.; Lee, J. Sparse Hierarchical Representation Learning on Functional Brain Networks for Prediction of Autism Severity Levels. Front. Neurosci. 2022, 16, 935431. [Google Scholar] [CrossRef]
- Schulte-Rüther, M.; Kulvicius, T.; Stroth, S.; Wolff, N.; Roessner, V.; Marschik, P.B.; Kamp-Becker, I.; Poustka, L. Using machine learning to improve diagnostic assessment of ASD in the light of specific differential and co-occurring diagnoses. J. Child Psychol. Psychiatry 2022, 64, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Jensen, M.; Amini, H.; Hormozdiari, F.; Penn, O.; Shifman, S.; Girirajan, S.; Hormozdiari, F. Dissecting the genetic basis of comorbid epilepsy phenotypes in neurodevelopmental disorders. Genome Med. 2019, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Gialloreti, L.E.; Enea, R.; Di Micco, V.; Di Giovanni, D.; Curatolo, P. Clustering analysis supports the detection of biological processes related to autism spectrum disorder. Genes 2020, 11, 1476. [Google Scholar] [CrossRef]
- Belmadani, M.; Jacobson, M.; Holmes, N.; Phan, M.; Nguyen, T.; Pavlidis, P.; Rogic, S. VariCarta: A Comprehensive Database of Harmonized Genomic Variants Found in Autism Spectrum Disorder Sequencing Studies. Autism Res. 2019, 12, 1728–1736. [Google Scholar] [CrossRef]
- Harris, Z.S. Distributional structure. Word 1954, 10, 146–162. [Google Scholar] [CrossRef]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: Distributed representation of genes based on co-expression. In Proceedings of the Selected Articles from the International Conference on Intelligent Biology and Medicine (ICIBM) 2018: Genomics, Los Angeles, CA, USA, 10–12 June 2018. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- The Gene Ontology and Consortium, Gene Ontology. Available online: http://geneontology.org (accessed on 18 May 2020).
- Řehůřek, R.; Sojka, P. Software Framework for Topic Modelling with Large Corpora. In Proceedings of the LREC 2010 Workshop on New Challenges for NLP Frameworks, Valletta, Malta, 22 May 2010. [Google Scholar]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A Tool for Annotating and Analyzing Human Hereditary Disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yao, H.; Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing 2016, 184, 232–242. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Maaten, L.V.D.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Dorrity, M.W.; Saunders, L.M.; Queitsch, C.; Fields, S.; Trapnell, C. Dimensionality reduction by UMAP to visualize physical and genetic interactions. Nat. Commun. 2020, 11, 1537. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Campello, R.J.G.B.; Moulavi, D.; Zimek, A.; Sander, J. Hierarchical Density Estimates for Data Clustering, Visualization, and Outlier Detection. ACM Trans. Knowl. Discov. Data 2015, 10, 5. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the KDD-96 Proceedings, Portland OR, USA, 2–4 August 1996. [Google Scholar]
- Parlett-Pelleriti, C.M.; Stevens, E.; Dixon, D.; Linstead, E.J. Applications of Unsupervised Machine Learning in Autism Spectrum Disorder Research: A Review. Rev. J. Autism Dev. Disord. 2022, 1–16. [Google Scholar] [CrossRef]
- Abdolzadegan, D.; Moattar, M.H.; Ghoshuni, M. A robust method for early diagnosis of autism spectrum disorder from EEG signals based on feature selection and DBSCAN method. Biocybern. Biomed. Eng. 2020, 40, 482–493. [Google Scholar] [CrossRef]
- Moulavi, D.; Jaskowiak, P.A.; Campello, R.J.G.B.; Zimek, A.; Sander, J. Density-Based Clustering Validation. In Proceedings of the 14th SIAM International Conference on Data Mining (SDM), Philadelphia, PA, USA, 24–26 April 2014. [Google Scholar]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Vesztrocy, A.W.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [Green Version]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An evolving database for the autism research community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef]
- Abrahams, B.; Arking, D.; Campbell, D.; Mefford, H.; Morrow, E.; Weiss, L.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Harrison, P.; Pattison, A.; Powell, D.; Beilharz, T. Topconfects: A package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biol. 2019, 20, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Nisar, S.; Hashem, S.; Bhat, A.; Syed, N.; Yadav, S.; Uddin, A.M.S.; Bagga, P.; Reddy, R.; Haris, M. Association of genes with phenotype in autism spectrum disorder. Aging 2019, 11, 10742–10770. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P.A. Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Bruno, L.; Doddato, G.; Valentino, F.; Baldassarri, M.; Tita, R.; Fallerini, C.; Bruttini, M.; Rizzo, C.L.; Mencarelli, M.A.; Mari, F.; et al. New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. Int. J. Mol. Sci. 2021, 22, 13439. [Google Scholar] [CrossRef]
- Ferhat, A.-T.; Halbedl, S.; Schmeisser, M.J.; Kas, M.J.; Bourgeron, T.; Ey, E. Behavioural Phenotypes and Neural Circuit Dysfunctions in Mouse Models of Autism Spectrum Disorder. Transl. Anat. Cell Biol. Autism Spectr. Disord. 2017, 224, 85–101. [Google Scholar]
- Muhle, R.; Reed, H.; Stratigos, K.; Veenstra-Vander Weele, J. The Emerging Clinical Neuroscience of Autism Spectrum Disorder: A Review. JAMA Psychiatry 2018, 75, 514–523. [Google Scholar] [CrossRef]
- Yang, Z.; Qing, H.; Gui, H.; Luo, J.; Dai, L.; Wang, B. Role of caprin-1 in carcinogenesis (Review). Oncol. Lett. 2019, 18, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Takao, K.; Miyakawa, T.; Shiina, N. Comprehensive behavioral analysis of 8 RNG105 (Caprin1) heterozygous mice: Reduced social interaction and attenuated response to 9 novelty. Sci. Rep. 2016, 6, 20775. [Google Scholar] [CrossRef] [PubMed]
- Pavinato, L.; Vedove, A.D.; Carli, D.; Ferrero, M.; Carestiato, S.; Howe, J.; Agolini, E.; Coviello, D.; van de Laar, I.; Au, P.Y.B.; et al. CAPRIN1 haploinsufficiency causes a neurodevelopmental disorder with language impairment, ADHD and ASD. Brain 2022, 2022, awac278. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ha, S.; Ahn, J.; Lee, S.; Choi, J.; Cheon, K. The Role of Ion Channel-Related Genes in Autism Spectrum Disorder: A Study Using Next-Generation Sequencing. Front. Genet. 2021, 12, 1935. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Yang, H.; Howrigan, D.; Wilkinson, B.; Souaiaia, T.; Evgrafov, O.; Genovese, G.; Clementel, V.; Tudor, J.; et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci. 2017, 20, 1150–1161. [Google Scholar] [CrossRef]
- Eisenberg, C.; Subramanian, D.; Afrasiabi, M.; Ziobro, P.; DeLucia, J.; Hirschberg, P.; Shiflett, M.; Santhakumar, V.; Tran, T. Reduced hippocampal inhibition and enhanced autism-epilepsy comorbidity in mice lacking neuropilin. Transl. Psychiatry 2021, 11, 537. [Google Scholar] [CrossRef]
- Yang, S.; Guo, X.; Dong, X.; Han, Y.; Gao, L.; Su, Y.; Dai, W.; Zhang, X. GABAA receptor subunit gene polymorphisms predict symptom-based and developmental deficits in Chinese Han children and adolescents with autistic spectrum disorders. Sci. Rep. 2017, 7, 3290. [Google Scholar] [CrossRef] [Green Version]
- Menzikov, S.; Morozov, S.; Kubatiev, A. Intricacies of GABAA Receptor Function: The Critical Role of the β3 Subunit in Norm and Pathology. Int. J. Mol. Sci. 2021, 22, 1457. [Google Scholar] [CrossRef]
- Zhang, X.; Piano, I.; Messina, A.; D’Antongiovanni, V.; Crò, F.; Provenzano, G.; Bozzi, Y.; Gargini, C.; Casarosa, S. Retinal defects in mice lacking the autism-associated gene Engrailed-2. Neuroscience 2019, 408, 177–190. [Google Scholar] [CrossRef]
- Cheng, N.; Pagtalunan, E.; Abushaibah, A.; Naidu, J.; Stell, W.K.; Rho, J.M.; Sauvé, Y. Atypical visual processing in a mouse model of autism. Sci. Rep. 2020, 10, 12390. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Go, Y.; Kushima, I.; Toyoda, A.; Fujiyama, A.; Imai, H.; Saito, N.; Iriki, A.; Ozaki, N.; Isoda, M. Single-neuron and genetic correlates of autistic behavior in macaque. Sci. Adv. 2016, 2, e1600558. [Google Scholar] [CrossRef] [PubMed]
- Ueoka, I.; Kawashima, H.; Konishi, A.; Aoki, M.; Tanaka, R.; Yoshida, H.; Maeda, T.; Ozaki, M.; Yamaguchi, M. Novel Drosophila model for psychiatric disorders including autism spectrum disorder by targeting of ATP-binding cassette protein A. Exp. Neurol. 2018, 300, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Ziats, C.; Patterson, W.; Friez, M. Syndromic Autism Revisited: Review of the Literature and Lessons Learned. Pediatr. Neurol. 2021, 114, 21–25. [Google Scholar] [CrossRef]
- Sztainberg, Y.Z.H. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Fernandez, B.; Scherer, S. Syndromic autism spectrum disorders: Moving from a clinically defined to a molecularly defined approach. Dialogues Clin. Neurosci. 2017, 19, 353–371. [Google Scholar] [CrossRef]
- Casanova, M.; Casanova, E.; Frye, R.; Baeza-Velasco, C.; LaSalle, J.; Hagerman, R.J.; Scherer, S.; Natowicz, M. Editorial: Secondary vs. Idiopathic Autism. Front. Psychiatry 2020, 11, 297. [Google Scholar] [CrossRef]
- Hughes, H.; Moreno, R.J.; Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain Behav. Immun. 2022, 108, 245–254. [Google Scholar] [CrossRef]
- Whiteley, P.; Marlow, B.; Kapoor, R.; Blagojevic-Stokic, N.; Sala, R. Autoimmune Encephalitis and Autism Spectrum Disorder. Front. Psychiatry 2021, 12, 775017. [Google Scholar] [CrossRef]
- Rowland, J.; Wilson, C. The association between gestational diabetes and ASD and ADHD: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 5136. [Google Scholar] [CrossRef] [PubMed]
- Gallego, B.; Walter, S.R.; Day, R.D.; Dunn, A.G.; Sivaraman, V.; Shah, N.; Longhurst, C.A.; Coiera, E. Bringing cohort studies to the bedside: Framework for a ‘green button’ to support clinical decision-making. J. ComEff. Res. 2015, 4, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, A.; Stein, G.; Ruppin, E.; Altman, R.; Sharan, R. A method for inferring medical diagnoses from patient similarities. BMC Med. 2013, 11, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wang, F.; Hu, J.; Sorrentino, R. Towards personalized medicine: Leveraging patient similarity and drug similarity analytics. AMIA Jt. Summits Transl. Sci. Proc. 2014, 2014, 132–136. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLUSTER INDEX | INDIVIDUALS | VARIANTS | ASD-LINKED GENES * |
|---|---|---|---|
| 0 | 1455 | 17,217 | 879 |
| 1 | 841 | 1747 | 154 |
| 2 | 273 | 7509 | 516 |
| 3 | 110 | 558 | 49 |
| 4 | 106 | 492 | 41 |
| 5 | 214 | 944 | 96 |
| 6 | 334 | 1859 | 188 |
| 7 | 336 | 5296 | 410 |
| 8 | 154 | 1186 | 117 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| molecular_function | GO:0005515 | Protein binding | 1.087222547 | 1.55 × 10−99 |
| cellular_component | GO:0005886 | Plasma membrane | 1.147275591 | 2.24 × 10−55 |
| cellular_component | GO:0005737 | Cytoplasm | 1.130739139 | 4.92 × 10−47 |
| cellular_component | GO:0005829 | Cytosol | 1.121747304 | 5.30 × 10−46 |
| molecular_function | GO:0005524 | ATP binding | 1.224015929 | 2.82 × 10−35 |
| molecular_function | GO:0046872 | Metal ion binding | 1.161264333 | 4.55 × 10−29 |
| cellular_component | GO:0005654 | Nucleoplasm | 1.116129667 | 2.34 × 10−27 |
| cellular_component | GO:0000786 | Nucleosome | 0.299454744 | 3.29 × 10−25 |
| cellular_component | GO:0005634 | Nucleus | 1.083234714 | 1.05 × 10−21 |
| cellular_component | GO:0005794 | Golgi apparatus | 1.20614718 | 1.76 × 10−20 |
| cellular_component | GO:0016020 | Membrane | 1.128252261 | 6.39 × 10−17 |
| molecular_function | GO:0004712 | Protein serine/threonine/tyrosine kinase activity | 1.290737468 | 1.18 × 10−16 |
| cellular_component | GO:0043231 | Intracellular membrane-bounded organelle | 1.198508723 | 3.19 × 10−16 |
| biological_process | GO:0006334 | Nucleosome assembly | 0.373973889 | 5.38 × 10−16 |
| cellular_component | GO:0005887 | Integral component of plasma membrane | 1.148641895 | 1.45 × 10−14 |
| molecular_function | GO:0004674 | Protein serine/threonine kinase activity | 1.29806618 | 1.69 × 10−14 |
| molecular_function | GO:0106310 | Protein serine kinase activity | 1.294450396 | 2.73 × 10−14 |
| cellular_component | GO:0098978 | Glutamatergic synapse | 1.297368237 | 2.94 × 10−13 |
| biological_process | GO:0006468 | Protein phosphorylation | 1.250748447 | 3.98 × 10−13 |
| cellular_component | GO:0030424 | Axon | 1.289923348 | 7.28 × 10−13 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| cellular_component | GO:0005886 | Plasma membrane | 1.246675801 | 1.52 × 10−4 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| biological_process | GO:0006939 | Smooth muscle contraction | 2.82253091 | 4.97 × 10−3 |
| molecular_function | GO:0005001 | Transmembrane receptor protein tyrosine phosphatase activity | 2.82253091 | 2.50 × 10−3 |
| cellular_component | GO:0016342 | Catenin complex | 2.763999163 | 8.48 × 10−7 |
| molecular_function | GO:1904315 | Transmitter-gated ion channel activity involved in regulation of postsynaptic membrane potential | 2.520116883 | 5.00 × 10−5 |
| biological_process | GO:0060078 | Regulation of postsynaptic membrane potential | 2.513396572 | 6.85 × 10−4 |
| cellular_component | GO:0044295 | Axonal growth cone | 2.492624699 | 1.75 × 10−3 |
| biological_process | GO:0098742 | Cell–cell adhesion via plasma-membrane adhesion molecules | 2.408414405 | 3.11 × 10−4 |
| molecular_function | GO:0043325 | Phosphatidylinositol-3,4-bisphosphate binding | 2.363695836 | 2.79 × 10−3 |
| cellular_component | GO:0099061 | Integral component of postsynaptic density membrane | 2.150499741 | 1.16 × 10−4 |
| cellular_component | GO:0098839 | Postsynaptic density membrane | 2.089853025 | 2.01 × 10−3 |
| biological_process | GO:0050804 | Modulation of chemical synaptic transmission | 2.068233856 | 1.14 × 10−3 |
| biological_process | GO:0051056 | Regulation of small GTPase-mediated signal transduction | 1.996809134 | 5.33 × 10−7 |
| molecular_function | GO:0008013 | β-catenin binding | 1.992650559 | 1.82 × 10−5 |
| cellular_component | GO:0031594 | Neuromuscular junction | 1.965691169 | 1.29 × 10−4 |
| cellular_component | GO:0098982 | GABA-ergic synapse | 1.943875232 | 1.98 × 10−4 |
| cellular_component | GO:0042734 | Presynaptic membrane | 1.924131347 | 1.54 × 10−3 |
| biological_process | GO:0043087 | Regulation of GTPase activity | 1.904088312 | 7.68 × 10−4 |
| biological_process | GO:0007411 | Axon guidance | 1.792469342 | 6.04 × 10−7 |
| biological_process | GO:0006470 | Protein dephosphorylation | 1.771967898 | 3.42 × 10−5 |
| cellular_component | GO:0045211 | Postsynaptic membrane | 1.764081818 | 9.26 × 10−6 |
| cellular_component | GO:0098685 | Schaffer collateral-CA1 synapse | 1.761281689 | 3.45 × 10−3 |
| cellular_component | GO:0098978 | Glutamatergic synapse | 1.713679481 | 3.42 × 10−12 |
| cellular_component | GO:0042383 | Sarcolemma | 1.660679084 | 3.34 × 10−3 |
| molecular_function | GO:0017124 | SH3 domain binding | 1.658399498 | 2.01 × 10−3 |
| biological_process | GO:0009887 | Animal organ morphogenesis | 1.65020987 | 2.70 × 10−3 |
| biological_process | GO:0007420 | Brain development | 1.628703639 | 4.48 × 10−6 |
| cellular_component | GO:0005938 | Cell cortex | 1.626910899 | 1.29 × 10−4 |
| biological_process | GO:0098609 | Cell–cell adhesion | 1.608369569 | 5.42 × 10−4 |
| cellular_component | GO:0005912 | Adherens junction | 1.603442789 | 1.16 × 10−4 |
| molecular_function | GO:0005085 | Guanyl nucleotide exchange factor activity | 1.591273804 | 2.63 × 10−5 |
| cellular_component | GO:0014069 | Postsynaptic density | 1.58515352 | 4.25 × 10−6 |
| biological_process | GO:0007268 | Chemical synaptic transmission | 1.574355946 | 4.57 × 10−5 |
| cellular_component | GO:0030054 | Cell junction | 1.556385229 | 7.80 × 10−5 |
| cellular_component | GO:0030424 | Axon | 1.555005455 | 1.13 × 10−7 |
| cellular_component | GO:0043005 | Neuron projection | 1.550471911 | 1.13 × 10−7 |
| biological_process | GO:0016477 | Cell migration | 1.545671689 | 1.07 × 10−4 |
| cellular_component | GO:0043197 | Dendritic spine | 1.54344642 | 2.41 × 10−3 |
| cellular_component | GO:0042995 | Cell projection | 1.542311533 | 3.45 × 10−3 |
| cellular_component | GO:0045202 | Synapse | 1.525092744 | 7.81 × 10−9 |
| cellular_component | GO:0030425 | Dendrite | 1.511341422 | 2.49 × 10−8 |
| molecular_function | GO:0005516 | Calmodulin binding | 1.508037943 | 2.77 × 10−3 |
| biological_process | GO:0007399 | Nervous system development | 1.502114113 | 4.57 × 10−5 |
| molecular_function | GO:0031267 | Small GTPase binding | 1.501876399 | 1.15 × 10−4 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| cellular_component | GO:0032391 | Photoreceptor connecting cilium | 8.843272901 | 1.02 × 10−3 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| molecular_function | GO:0005516 | Calmodulin binding | 2.394767442 | 9.63 × 10−4 |
| cellular_component | GO:0030424 | Axon | 1.983969128 | 2.02 × 10−3 |
| molecular_function | GO:0005524 | ATP binding | 1.548471524 | 1.05 × 10−5 |
| cellular_component | GO:0005886 | Plasma membrane | 1.271435899 | 5.21 × 10−6 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| molecular_function | GO:0008066 | Glutamate receptor activity | 4.18111949 | 2.10 × 10−5 |
| biological_process | GO:0007413 | Axonal fasciculation | 3.520942728 | 2.12 × 10−3 |
| molecular_function | GO:0098632 | Cell–cell adhesion mediator activity | 3.185614849 | 2.38 × 10−4 |
| molecular_function | GO:0050840 | Extracellular matrix binding | 3.026334107 | 3.24 × 10−4 |
| cellular_component | GO:0016342 | Catenin complex | 2.774567772 | 1.13 × 10−3 |
| biological_process | GO:0050804 | Modulation of chemical synaptic transmission | 2.553984319 | 3.06 × 10−4 |
| cellular_component | GO:0099061 | Integral component of postsynaptic density membrane | 2.248669305 | 3.79 × 10−3 |
| cellular_component | GO:0005912 | Adherents junction | 2.123743233 | 2.53 × 10−9 |
| biological_process | GO:0051056 | Regulation of small GTPase-mediated signal transduction | 1.994471906 | 1.25 × 10−3 |
| biological_process | GO:0018108 | Peptidyl-tyrosine phosphorylation | 1.978565473 | 6.13 × 10−4 |
| biological_process | GO:0007411 | Axon guidance | 1.894891591 | 2.12 × 10−4 |
| cellular_component | GO:0030424 | Axon | 1.858275329 | 9.42 × 10−11 |
| molecular_function | GO:0005201 | Extracellular matrix structural constituent | 1.848586719 | 9.09 × 10−4 |
| molecular_function | GO:0008017 | Microtubule binding | 1.810574065 | 1.12 × 10−6 |
| cellular_component | GO:0045211 | Postsynaptic membrane | 1.791908353 | 1.55 × 10−3 |
| biological_process | GO:0007156 | Homophilic cell adhesion via plasma membrane adhesion molecules | 1.784711934 | 2.12 × 10−3 |
| molecular_function | GO:0051015 | Actin filament binding | 1.73356715 | 1.40 × 10−4 |
| molecular_function | GO:0005516 | Calmodulin binding | 1.720232019 | 3.71 × 10−4 |
| molecular_function | GO:0003779 | Actin binding | 1.719508015 | 1.91 × 10−5 |
| cellular_component | GO:0098978 | Glutamatergic synapse | 1.643533776 | 8.85 × 10−6 |
| cellular_component | GO:0043235 | Receptor complex | 1.637052075 | 8.44 × 10−4 |
| biological_process | GO:0007420 | Brain development | 1.61913482 | 4.92 × 10−3 |
| molecular_function | GO:0005096 | GTPase activator activity | 1.598663334 | 5.70 × 10−4 |
| GO Element Type | GO Code | GO Name | FE | FDR |
|---|---|---|---|---|
| molecular_function | GO:0004712 | Protein serine/threonine/tyrosine kinase activity | 2.162900762 | 1.88 × 10−3 |
| molecular_function | GO:0005524 | ATP binding | 1.716714944 | 1.24 × 10−5 |
| Cluster/ Phenotype | 0 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Restrictive behavior | 1.22 | 2.33 | 1.23 | 1.93 | ||||
| Impaired social interactions | 1.05 | 1.16 | 1.12 | 3.81 | 1.32 | 2.01 | 1.18 | |
| Poor eye contact | 1.55 | 1.15 | 1.40 | 2.56 | ||||
| Lack of peer relationships | 1.20 | 1.65 | 4.16 | 2.22 | 1.17 | 1.74 | ||
| Restrictive behavior | 1.23 | |||||||
| Impaired ability to form peer relationships | 1.20 | 1.83 | 13.95 | |||||
| Abnormal non-verbal communicative behavior | 1.10 | 8.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Giovanni, D.; Enea, R.; Di Micco, V.; Benvenuto, A.; Curatolo, P.; Emberti Gialloreti, L. Using Machine Learning to Explore Shared Genetic Pathways and Possible Endophenotypes in Autism Spectrum Disorder. Genes 2023, 14, 313. https://doi.org/10.3390/genes14020313
Di Giovanni D, Enea R, Di Micco V, Benvenuto A, Curatolo P, Emberti Gialloreti L. Using Machine Learning to Explore Shared Genetic Pathways and Possible Endophenotypes in Autism Spectrum Disorder. Genes. 2023; 14(2):313. https://doi.org/10.3390/genes14020313
Chicago/Turabian StyleDi Giovanni, Daniele, Roberto Enea, Valentina Di Micco, Arianna Benvenuto, Paolo Curatolo, and Leonardo Emberti Gialloreti. 2023. "Using Machine Learning to Explore Shared Genetic Pathways and Possible Endophenotypes in Autism Spectrum Disorder" Genes 14, no. 2: 313. https://doi.org/10.3390/genes14020313