
Palmitic Acid Exerts Anti-Tumorigenic Activities by Modulating Cellular Stress and Lipid Droplet Formation in Endometrial Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Preparation of BSA-Bound PA
2.3. Primary Culture of Human-Derived EC
2.4. Lkb1fl/flp53fl/fl Transgenic Mouse Model of EC
2.5. Cell Viability Assay
2.6. Colony Assays
2.7. Preparation of Cell-Based Functional Assays
2.8. Glucose Uptake Assay
2.9. Lactate Assay
2.10. ATP Assay
2.11. Cleaved Caspase 3, 8, and 9 ELISA Assays
2.12. Reactive Oxygen Species (ROS) Assay
2.13. Mitochondrial Membrane Potential Assays
2.14. Adhesion Assay
2.15. Transwell Assay
2.16. Wound Healing Assay
2.17. Fatty Acid Oxidation (FAO) Assay
2.18. Oil Red O Staining Assay
2.19. Western Immunoblotting
2.20. Immunohistochemistry (IHC)
2.21. HE Staining
2.22. Serum Vascular Endothelial Growth Factor (VEGF) and Triglyceride (TG) Assays
2.23. Statistical Analysis
3. Results
3.1. PA Inhibited Cell Proliferation and the AKT/mTOR, MAPK, and p38 Pathways in EC Cells
3.2. PA Induced Apoptosis in EC Cells
3.3. PA Induced Cellular Stress in EC Cells
3.4. PA Reduced Adhesion, Migration, and Invasion in EC Cells
3.5. Cellular Stress Modulated PA-Induced Migration and Invasion
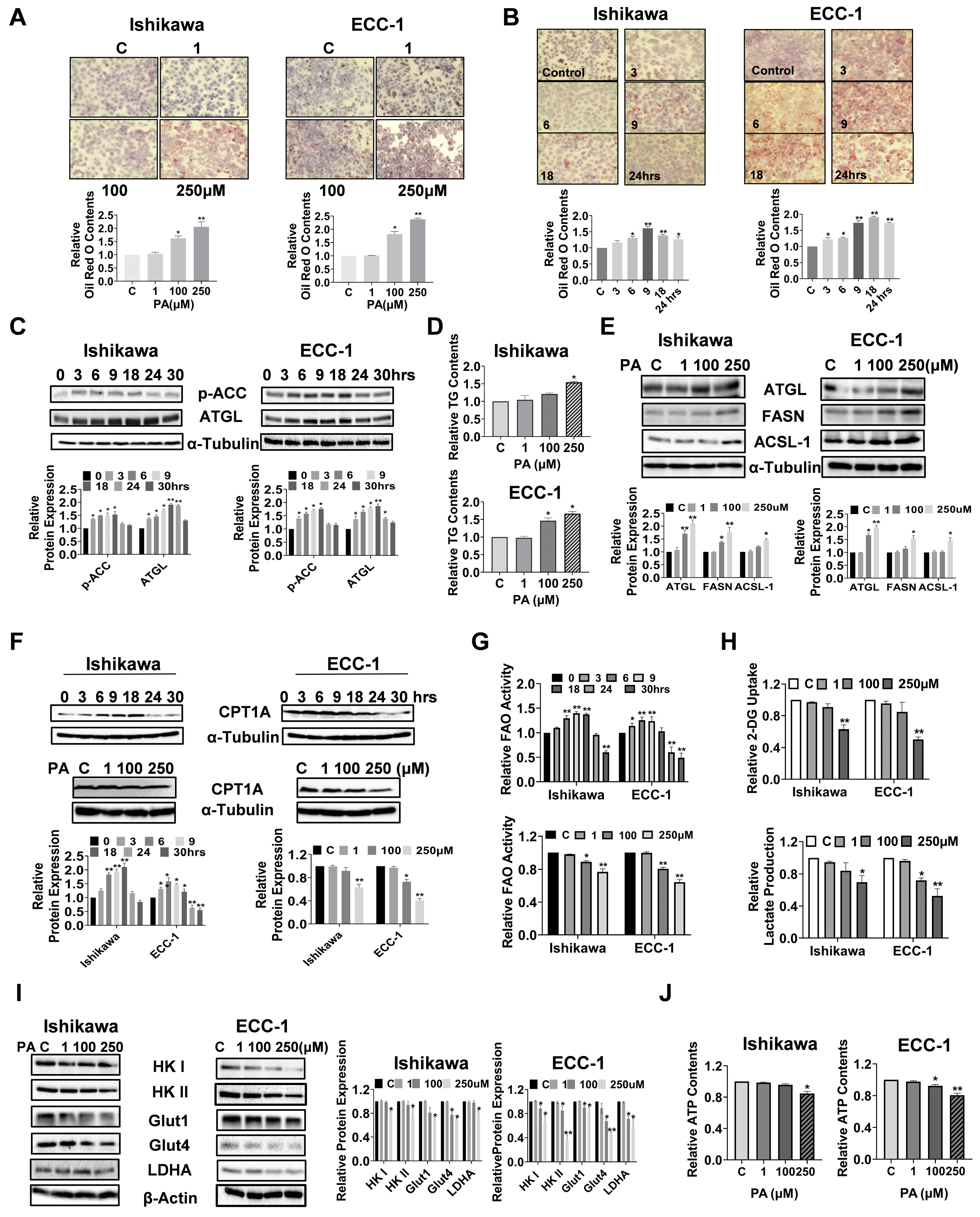
3.6. PA Caused Lipid Accumulation and Increased Lipogenesis in EC Cells
3.7. Inhibition of LD Formation Increased the Sensitivity to PA in EC Cells
3.8. PA Inhibited Tumor Growth and Increased Lipogenesis in the Lkb1fl/flp53fl/fl Mouse Mode of EC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Huang, Y.; Hou, J.Y.; Khoury-Collado, F.; Melamed, A.; St Clair, C.M.; Tergas, A.I.; Hur, C.; Hillyer, G.C.; Hershman, D.L.; et al. Toxicity after adjuvant therapy for stage III uterine cancer. Gynecol. Oncol. 2020, 159, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the Role of Obesity in Endometrial Cancer Risk, Prevention, and Treatment. J. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, K.; Niklas, N.; Rychlicka, M.; Cymbaluk-Płoska, A. The Influence of Biologically Active Substances Secreted by the Adipose Tissue on Endometrial Cancer. Diagnostics 2021, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.A.; Holland, C.M.; Kreil, D.P.; MacKay, D.J.C.; Charnock-Jones, D.S.; Print, C.G.; Smith, S.K. Independent component analysis of microarray data in the study of endometrial cancer. Oncogene 2004, 23, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Njoku, K.; Sutton, C.J.J.; Whetton, A.D.; Crosbie, E.J. Metabolomic Biomarkers for Detection, Prognosis and Identifying Recurrence in Endometrial Cancer. Metabolites 2020, 10, 314. [Google Scholar] [CrossRef]
- Altadill, T.; Dowdy, T.M.; Gill, K.; Reques, A.; Menon, S.S.; Moiola, C.P.; Lopez-Gil, C.; Coll, E.; Matias-Guiu, X.; Cabrera, S.; et al. Metabolomic and Lipidomic Profiling Identifies The Role of the RNA Editing Pathway in Endometrial Carcinogenesis. Sci. Rep. 2017, 7, 8803. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Gatius, S.; Yeramian, A.; Portero-Otin, M.; Eritja, N.; Santacana, M.; Colas, E.; Ruiz, M.; Pamplona, R.; Matias-Guiu, X. Metabotyping human endometrioid endometrial adenocarcinoma reveals an implication of endocannabinoid metabolism. Oncotarget 2016, 7, 52364–52374. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- de Carvalho, C.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, M.A.; Pereira, L.; Fraile, C.; Valerio, L.; Balboa, M.A.; Balsinde, J. Roles of Palmitoleic Acid and Its Positional Isomers, Hypogeic and Sapienic Acids, in Inflammation, Metabolic Diseases and Cancer. Cells 2022, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Feron, O. Dealing with saturated and unsaturated fatty acid metabolism for anticancer therapy. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; Cohen, O.; Daniel, B.; Ferreri, C.; Sasson, S. The Combination of Whole Cell Lipidomics Analysis and Single Cell Confocal Imaging of Fluidity and Micropolarity Provides Insight into Stress-Induced Lipid Turnover in Subcellular Organelles of Pancreatic Beta Cells. Molecules 2019, 24, 3742. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Sansone, A.; Ferreri, R.; Amézaga, J.; Tueros, I. Fatty Acids and Membrane Lipidomics in Oncology: A Cross-Road of Nutritional, Signaling and Metabolic Pathways. Metabolites 2020, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016, 7, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Hu, X.; Gong, R.H.; Huang, C.; Chen, M.; Wong, H.L.X.; Bian, Z.; Kwan, H.Y. Palmitic acid is an intracellular signaling molecule involved in disease development. Cell. Mol. Life Sci. 2019, 76, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Murru, E.; Manca, C.; Carta, G.; Banni, S. Impact of Dietary Palmitic Acid on Lipid Metabolism. Front. Nutr. 2022, 9, 861664. [Google Scholar] [CrossRef]
- Waki, M.; Ide, Y.; Ishizaki, I.; Nagata, Y.; Masaki, N.; Sugiyama, E.; Kurabe, N.; Nicolaescu, D.; Yamazaki, F.; Hayasaka, T.; et al. Single-cell time-of-flight secondary ion mass spectrometry reveals that human breast cancer stem cells have significantly lower content of palmitoleic acid compared to their counterpart non-stem cancer cells. Biochimie 2014, 107, 73–77. [Google Scholar] [CrossRef]
- Igal, R.A. Roles of StearoylCoA Desaturase-1 in the Regulation of Cancer Cell Growth, Survival and Tumorigenesis. Cancers 2011, 3, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Koltun, D.O.; Vasilevich, N.I.; Parkhill, E.Q.; Glushkov, A.I.; Zilbershtein, T.M.; Mayboroda, E.I.; Boze, M.A.; Cole, A.G.; Henderson, I.; Zautke, N.A.; et al. Orally bioavailable, liver-selective stearoyl-CoA desaturase (SCD) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3050–3053. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Fatima, S.; Chen, M.; Xu, K.; Huang, C.; Gong, R.H.; Su, T.; Wong, H.L.X.; Bian, Z.; Kwan, H.Y. Toll-like receptor 4 is a master regulator for colorectal cancer growth under high-fat diet by programming cancer metabolism. Cell Death Dis. 2021, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Hu, X.; Huang, C.; Zhang, W.; Cai, J.; Huang, M.; Gong, R.H.; Chen, M.; Ho, A.H.M.; Su, T.; et al. High-fat diet feeding and palmitic acid increase CRC growth in β2AR-dependent manner. Cell Death Dis. 2019, 10, 711. [Google Scholar] [CrossRef] [PubMed]
- Bojková, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary Fat and Cancer-Which Is Good, Which Is Bad, and the Body of Evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef] [PubMed]
- Sczaniecka, A.K.; Brasky, T.M.; Lampe, J.W.; Patterson, R.E.; White, E. Dietary intake of specific fatty acids and breast cancer risk among postmenopausal women in the VITAL cohort. Nutr. Cancer 2012, 64, 1131–1142. [Google Scholar] [CrossRef]
- Li, L.; Zeng, X.; Liu, Z.; Chen, X.; Li, L.; Luo, R.; Liu, X.; Zhang, J.; Liu, J.; Lu, Y.; et al. Mesenchymal stromal cells protect hepatocytes from lipotoxicity through alleviation of endoplasmic reticulum stress by restoring SERCA activity. J. Cell. Mol. Med. 2021, 25, 2976–2993. [Google Scholar] [CrossRef]
- Bolognesi, A.; Chatgilialoglu, A.; Polito, L.; Ferreri, C. Membrane lipidome reorganization correlates with the fate of neuroblastoma cells supplemented with fatty acids. PLoS ONE 2013, 8, e55537. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Ferreri, R.; Amézaga, J.; Burgos, M.C.; Arranz, S.; Tueros, I. Critical Review on Fatty Acid-Based Food and Nutraceuticals as Supporting Therapy in Cancer. Int. J. Mol. Sci. 2022, 23, 6030. [Google Scholar] [CrossRef]
- Cousin, S.P.; Hügl, S.R.; Wrede, C.E.; Kajio, H.; Myers, M.G., Jr.; Rhodes, C.J. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology 2001, 142, 229–240. [Google Scholar] [CrossRef]
- Guo, H.; Kong, W.; Zhang, L.; Han, J.; Clark, L.H.; Yin, Y.; Fang, Z.; Sun, W.; Wang, J.; Gilliam, T.P.; et al. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am. J. Cancer Res. 2019, 9, 2170–2193. [Google Scholar] [PubMed]
- Lin, L.; Ding, Y.; Wang, Y.; Wang, Z.; Yin, X.; Yan, G.; Zhang, L.; Yang, P.; Shen, H. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology 2017, 66, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, Y.; Han, D.; Lu, J.; Yin, S.; Hu, H.; Zhao, C. Combination of Palmitic Acid and Methylseleninic Acid Induces Mitochondria-Dependent Apoptosis via Attenuation of the IRE1α Arm and Enhancement of CHOP in Hepatoma. ACS Omega 2021, 6, 15708–15715. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jiao, W.; Xu, Y.; Hou, L.; Li, H.; Shao, J.; Zhang, X.; Wang, R.; Kong, D. Palmitic acid inhibits prostate cancer cell proliferation and metastasis by suppressing the PI3K/Akt pathway. Life Sci. 2021, 286, 120046. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Li, C.; Li, L.; Lian, J.; Watts, R.; Nelson, R.; Goodwin, B.; Lehner, R. Roles of Acyl-CoA:Diacylglycerol Acyltransferases 1 and 2 in Triacylglycerol Synthesis and Secretion in Primary Hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing fatty acid-induced lipotoxicity and its therapeutic potential in glioblastoma using stimulated Raman microscopy. Sci. Rep. 2021, 11, 7422. [Google Scholar] [CrossRef] [PubMed]
- Mozihim, A.K.; Chung, I.; Said, N.; Jamil, A.H.A. Reprogramming of Fatty Acid Metabolism in Gynaecological Cancers: Is There a Role for Oestradiol? Metabolites 2022, 12, 350. [Google Scholar] [CrossRef]
- de Araujo Junior, R.F.; Eich, C.; Jorquera, C.; Schomann, T.; Baldazzi, F.; Chan, A.B.; Cruz, L.J. Ceramide and palmitic acid inhibit macrophage-mediated epithelial-mesenchymal transition in colorectal cancer. Mol. Cell. Biochem. 2020, 468, 153–168. [Google Scholar] [CrossRef]
- Makker, A.; Goel, M.M. Tumor progression, metastasis, and modulators of epithelial-mesenchymal transition in endometrioid endometrial carcinoma: An update. Endocr.-Relat. Cancer 2016, 23, R85–R111. [Google Scholar] [CrossRef] [PubMed]
- Colas, E.; Pedrola, N.; Devis, L.; Ertekin, T.; Campoy, I.; Martínez, E.; Llauradó, M.; Rigau, M.; Olivan, M.; Garcia, M.; et al. The EMT signaling pathways in endometrial carcinoma. Clin. Transl. Oncol. 2012, 14, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Lei, H.; Yi, T.; Su, P.; Tang, S.; Tong, Y.; Dong, B.; Ruan, G.; Mustea, A.; Sehouli, J.; et al. Lipid reprogramming induced by the TFEB-ERRα axis enhanced membrane fluidity to promote EC progression. J. Exp. Clin. Cancer Res. 2022, 41, 28. [Google Scholar] [CrossRef] [PubMed]
- Troisi, J.; Sarno, L.; Landolfi, A.; Scala, G.; Martinelli, P.; Venturella, R.; Di Cello, A.; Zullo, F.; Guida, M. Metabolomic Signature of Endometrial Cancer. J. Proteome Res. 2018, 17, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yu, X.; Peng, C.; Liu, N.; Chen, W.; Xu, H.; Wei, H.; Fang, K.; Dong, Z.; Fu, C.; et al. Activation of SREBP-1c alters lipogenesis and promotes tumor growth and metastasis in gastric cancer. Biomed. Pharmacother. 2020, 128, 110274. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Hofmann, W.A. Effect of Palmitic Acid on Exosome-Mediated Secretion and Invasive Motility in Prostate Cancer Cells. Molecules 2020, 25, 2722. [Google Scholar] [CrossRef] [PubMed]
- Landim, B.C.; de Jesus, M.M.; Bosque, B.P.; Zanon, R.G.; da Silva, C.V.; Góes, R.M.; Ribeiro, D.L. Stimulating effect of palmitate and insulin on cell migration and proliferation in PNT1A and PC3 prostate cells: Counteracting role of metformin. Prostate 2018, 78, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Farhat, D.; Ghayad, S.E.; Icard, P.; Le Romancer, M.; Hussein, N.; Lincet, H. Lipoic acid-induced oxidative stress abrogates IGF-1R maturation by inhibiting the CREB/furin axis in breast cancer cell lines. Oncogene 2020, 39, 3604–3610. [Google Scholar] [CrossRef]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Pagliassotti, M.J.; Kim, P.Y.; Estrada, A.L.; Stewart, C.M.; Gentile, C.L. Endoplasmic reticulum stress in obesity and obesity-related disorders: An expanded view. Metabolism 2016, 65, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Rizzo, A.M.; Colombo, I.; Montorfano, G.; Zava, S.; Corsetto, P.A. Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 2021, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Lei, L.; Lv, Q.; Gong, Y.; Yang, L. Curcumin attenuates palmitic acid-induced cell apoptosis by inhibiting endoplasmic reticulum stress in H9C2 cardiomyocytes. Hum. Exp. Toxicol. 2019, 38, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, D.; Wang, F.; Wang, C.; Yang, L. Curcumin protects against palmitic acid-induced apoptosis via the inhibition of endoplasmic reticulum stress in testicular Leydig cells. Reprod. Biol. Endocrinol. 2019, 17, 71. [Google Scholar] [CrossRef]
- Xue, X.; Li, F.; Cai, M.; Hu, J.; Wang, Q.; Lou, S. Interactions between Endoplasmic Reticulum Stress and Autophagy: Implications for Apoptosis and Neuroplasticity-Related Proteins in Palmitic Acid-Treated Prefrontal Cells. Neural Plast. 2021, 2021, 8851327. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Luo, H.; Zhang, N.; Wang, Y.; Li, Y.; Huang, H.; Liu, Y.; Hu, Y.; Liu, H.; Zhang, J.; et al. Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2019, 20, 6268. [Google Scholar] [CrossRef]
- Pardo, V.; González-Rodríguez, Á.; Muntané, J.; Kozma, S.C.; Valverde, Á.M. Role of hepatocyte S6K1 in palmitic acid-induced endoplasmic reticulum stress, lipotoxicity, insulin resistance and in oleic acid-induced protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef]
- Chen, Y.P.; Kuo, W.W.; Baskaran, R.; Day, C.H.; Chen, R.J.; Wen, S.Y.; Ho, T.J.; Padma, V.V.; Kuo, C.H.; Huang, C.Y. Acute hypoxic preconditioning prevents palmitic acid-induced cardiomyocyte apoptosis via switching metabolic GLUT4-glucose pathway back to CD36-fatty acid dependent. J. Cell. Biochem. 2018, 119, 3363–3372. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhou, C.; Guo, X.; Ding, Y.; Ma, S.; Gong, X.; Jiang, H.; Wang, Y.; Wang, X. Palmitate impairs the autophagic flux to induce p62-dependent apoptosis through the upregulation of CYLD in NRCMs. Toxicology 2022, 465, 153032. [Google Scholar] [CrossRef]
- Li, S.; Sun, C.; Gu, Y.; Gao, X.; Zhao, Y.; Yuan, Y.; Zhang, F.; Hu, P.; Liang, W.; Cao, K.; et al. Mutation of IDH1 aggravates the fatty acid-induced oxidative stress in HCT116 cells by affecting the mitochondrial respiratory chain. Mol. Med. Rep. 2019, 19, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, L.; Guo, H.; Wysham, W.Z.; Roque, D.R.; Willson, A.K.; Sheng, X.; Zhou, C.; Bae-Jump, V.L. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol. Oncol. 2015, 138, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, H.; Ren, L.; Lu, Z.; Zheng, Q.; Ding, L.; Xie, H.; Wang, R.; Yu, C.; Lin, Y.; et al. Adding fuel to the fire: The lipid droplet and its associated proteins in cancer progression. Int. J. Biol. Sci. 2022, 18, 6020–6034. [Google Scholar] [CrossRef]
- Li, S.; Wu, T.; Lu, Y.X.; Wang, J.X.; Yu, F.H.; Yang, M.Z.; Huang, Y.J.; Li, Z.J.; Wang, S.L.; Huang, L.; et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020, 36, 101596. [Google Scholar] [CrossRef]
- Eynaudi, A.; Díaz-Castro, F.; Bórquez, J.C.; Bravo-Sagua, R.; Parra, V.; Troncoso, R. Differential Effects of Oleic and Palmitic Acids on Lipid Droplet-Mitochondria Interaction in the Hepatic Cell Line HepG2. Front. Nutr. 2021, 8, 775382. [Google Scholar] [CrossRef] [PubMed]
- Paramitha, P.N.; Zakaria, R.; Maryani, A.; Kusaka, Y.; Andriana, B.B.; Hashimoto, K.; Nakazawa, H.; Kato, S.; Sato, H. Raman Study on Lipid Droplets in Hepatic Cells Co-Cultured with Fatty Acids. Int. J. Mol. Sci. 2021, 22, 7378. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.; Obiang-Obounou, B.W.; Kim, M.; Choi, Y.; Lee, H.S.; Lee, K. Therapeutic strategies for metabolic diseases: Small-molecule diacylglycerol acyltransferase (DGAT) inhibitors. ChemMedChem 2014, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Kong, W.; Shen, X.; Han, C.; Zhao, Z.; Chen, S.; Zhou, C.; Bae-Jump, V. The role of DGAT1 and DGAT2 in regulating tumor cell growth and their potential clinical implications. J. Transl. Med. 2024, 22, 290. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Corbacho, M.J.; Obeid, L.M. A novel role for DGATs in cancer. Adv. Biol. Regul. 2019, 72, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B., Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 631. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Ochoa, E.; Sánchez-Alegría, K.; Gómez-Inclán, C.; Ferrera, P.; Arias, C. Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: The role of mTOR activation and mitochondrial ROS production. Neurochem. Int. 2017, 110, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Wu, Y.; Deol, P.; Nobumori, Y.; Zhou, Q.; Sladek, F.M.; Liu, X. Palmitic acid negatively regulates tumor suppressor PTEN through T366 phosphorylation and protein degradation. Cancer Lett. 2021, 496, 127–133. [Google Scholar] [CrossRef]
- Wang, T.; Yao, W.; Li, J.; He, Q.; Shao, Y.; Huang, F. Acetyl-CoA from inflammation-induced fatty acids oxidation promotes hepatic malate-aspartate shuttle activity and glycolysis. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E496–E510. [Google Scholar] [CrossRef]
- Wu, Z.S.; Huang, S.M.; Wang, Y.C. Palmitate Enhances the Efficacy of Cisplatin and Doxorubicin against Human Endometrial Carcinoma Cells. Int. J. Mol. Sci. 2021, 23, 80. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Chen, X.; Sun, Y.; Wang, L.; Shu, H.; Qian, C. A metabolomic signature of FIGO stage I and II endometrial cancer. Neoplasma 2021, 68, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, C.; Bao, N. Molecular mechanism of palmitic acid and its derivatives in tumor progression. Front. Oncol. 2023, 13, 1224125. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; de Araújo Júnior, R.F.; Cavalcante, R.S.; Yu, Z.; Schomann, T.; Gu, Z.; Eich, C.; Cruz, L.J. Effective breast cancer therapy based on palmitic acid-loaded PLGA nanoparticles. Biomater. Adv. 2023, 145, 213270. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Wang, J.; Kong, W.; Newton, M.A.; Burkett, W.C.; Sun, W.; Buckingham, L.; O’Donnell, J.; Suo, H.; Deng, B.; et al. Palmitic Acid Exerts Anti-Tumorigenic Activities by Modulating Cellular Stress and Lipid Droplet Formation in Endometrial Cancer. Biomolecules 2024, 14, 601. https://doi.org/10.3390/biom14050601
Zhao Z, Wang J, Kong W, Newton MA, Burkett WC, Sun W, Buckingham L, O’Donnell J, Suo H, Deng B, et al. Palmitic Acid Exerts Anti-Tumorigenic Activities by Modulating Cellular Stress and Lipid Droplet Formation in Endometrial Cancer. Biomolecules. 2024; 14(5):601. https://doi.org/10.3390/biom14050601
Chicago/Turabian StyleZhao, Ziyi, Jiandong Wang, Weimin Kong, Meredith A. Newton, Wesley C. Burkett, Wenchuan Sun, Lindsey Buckingham, Jillian O’Donnell, Hongyan Suo, Boer Deng, and et al. 2024. "Palmitic Acid Exerts Anti-Tumorigenic Activities by Modulating Cellular Stress and Lipid Droplet Formation in Endometrial Cancer" Biomolecules 14, no. 5: 601. https://doi.org/10.3390/biom14050601