


Genes 2024, 15(5), 609; https://doi.org/10.3390/genes15050609 (registering DOI) - 10 May 2024
Abstract
Schizophrenia symptomatology includes negative symptoms and cognitive impairment. Several studies have linked schizophrenia with the PDE4 family of enzymes due to their genetic association and function in cognitive processes such as long-term potentiation. We conducted a systematic gene expression meta-analysis of four PDE4
[...] Read more.
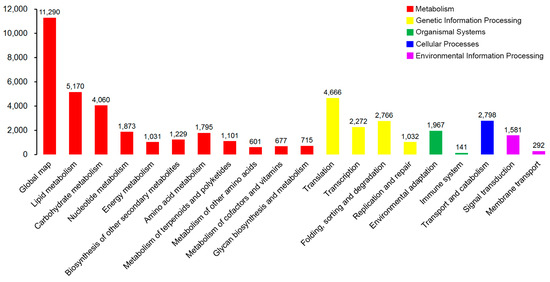
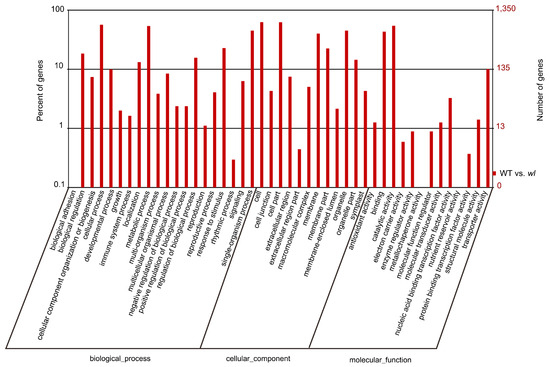
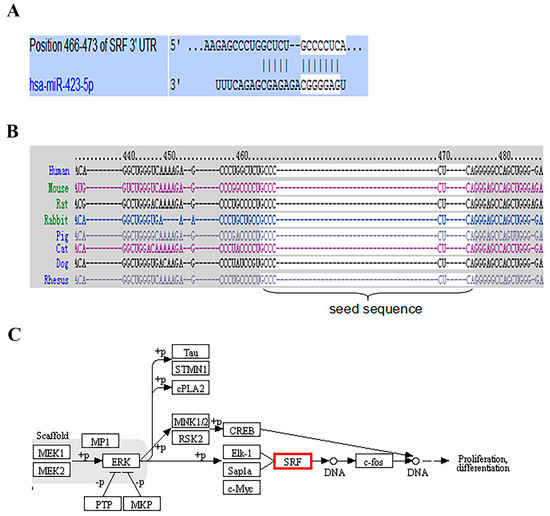
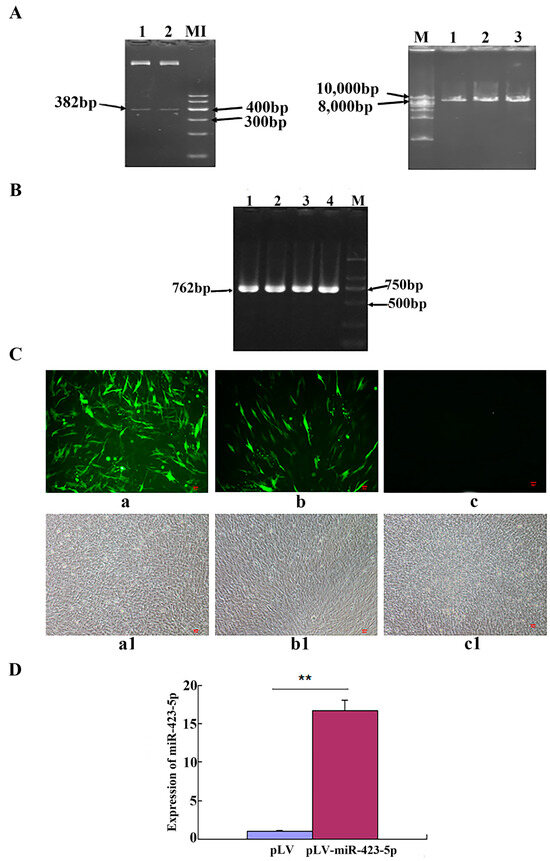
Schizophrenia symptomatology includes negative symptoms and cognitive impairment. Several studies have linked schizophrenia with the PDE4 family of enzymes due to their genetic association and function in cognitive processes such as long-term potentiation. We conducted a systematic gene expression meta-analysis of four PDE4 genes (PDE4A-D) in 10 brain sample datasets (437 samples) and three blood sample datasets (300 samples). Subsequently, we measured mRNA levels in iPSC-derived hippocampal dentate gyrus neurons generated from fibroblasts of three groups: healthy controls, healthy monozygotic twins (MZ), and their MZ siblings with schizophrenia. We found downregulation of PDE4B in brain tissues, further validated by independent data of the CommonMind consortium (515 samples). Interestingly, the downregulation signal was present in a subgroup of the patients, while the others showed no differential expression or even upregulation. Notably, PDE4A, PDE4B, and PDE4D exhibited upregulation in iPSC-derived neurons compared to healthy controls, whereas in blood samples, PDE4B was found to be upregulated while PDE4A was downregulated. While the precise mechanism and direction of altered PDE4 expression necessitate further investigation, the observed multilevel differential expression across the brain, blood, and iPSC-derived neurons compellingly suggests the involvement of PDE4 genes in the pathophysiology of schizophrenia.
Full article
(This article belongs to the Special Issue Genetic Basis Underlying Neuropsychiatric Disorders 2.0)
►
Show Figures
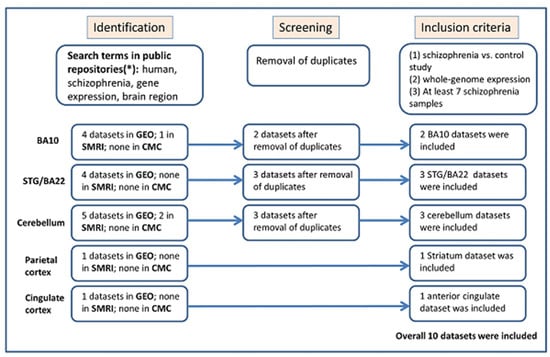
Figure 1


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}