
Divergent Synthesis of 5,7-Diazaullazines Derivatives through a Combination of Cycloisomerization with Povarov or Alkyne–Carbonyl Metathesis

Abstract
:
1. Introduction
1.1. Synthesis
1.2. Photophysical Properties
2. Conclusions
3. Materials and Methods
3.1. General Information
3.2. Analytical Data
3.2.1. General Procedure A for the Synthesis of 5,13-Diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5a–l)
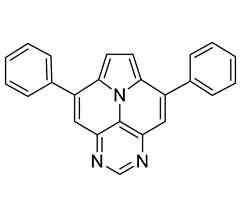
- 5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5a)
![Molecules 29 02159 i004]()
- According to general procedure A, the title compound 5a was obtained as an orange solid in 45% yield (54 mg, 0.121 mmol). Rf 0.50 (CH2Cl2/EtOAc 10:1). Mp. 284–286 °C. 1H NMR (500 MHz, CDCl3) δ = 8.83 (s, 1H), 8.31–8.27 (m, 1H), 8.24 (d, J = 4.3 Hz, 1H), 7.89–7.86 (m, 2H), 7.84 (ddd, J = 8.3 Hz, J = 6.6 Hz, J = 1.4 Hz, 1H), 7.66 (dd, J = 8.6 Hz, J = 1.4 Hz, 1H), 7.63–7.54 (m, 7H), 7.46 (ddd, J = 8.3 Hz, J = 6.7 Hz, J = 1.2 Hz, 1H), 7.42 (d, J = 4.3 Hz, 1H), 7.40–7.37 (m, 2H). 13C NMR (126 MHz, CDCl3) δ = 153.4, 150.2, 149.6, 146.9, 146.8, 144.5, 140.9, 138.7, 137.3, 131.5, 129.4, 129.1, 128.9, 128.7, 128.2, 127.9, 127.6, 127.5, 127.2, 126.3, 126.1, 122.1, 117.5, 116.8, 112.2, 109.1. IR (ATR, cm−1): ṽ = 1605 (s), 1578 (m), 1500 (m), 1449 (m), 1405 (m), 863 (m), 777 (s), 734 (s), 705 (vs), 612 (s), 554 (s). MS (EI, 70 eV): m/z (%) = 446 (86, M+), 445 (92), 444 (28), 382 (56), 381 (100), 380 (89), 379 (28), 354 (26), 223 (49), 222 (52), 208 (31). HRMS (ESI-TOF): calculated for C31H19N4 ([M + H]+) 447.1609, found 447.1602.
- 5,13-di-p-tolylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5b)
![Molecules 29 02159 i005]()
- According to general procedure A, the title compound 5b was obtained as an orange solid in 42% yield (50 mg, 0.105 mmol). Rf 0.78 (CH2Cl2/EtOAc 15:1). Mp. 303–306 °C. 1H NMR (250 MHz, CDCl3) δ = 8.83 (s, 1H), 8.22–8.16 (m, 1H), 8.13 (d, J = 4.3 Hz, 1H), 7.79–7.69 (m, 3H), 7.64 (ddd, J = 8.7 Hz, J = 1.5 Hz, J = 0.7 Hz, 1H), 7.49 (s, 1H), 7.44–7.35 (m, 5H), 7.33 (d, J = 4.3 Hz, 1H), 7.32–7.21 (m, 2H), 2.60 (s, 3H), 2.48 (s, 3H).13C NMR (63 MHz, CDCl3) δ = 153.3, 150.4, 149.4, 146.8, 146.7, 144.4, 140.7, 139.5, 137.0, 135.6, 134.4, 131.3, 129.7, 128.8, 128.8, 128.6, 128.5, 127.9, 127.4, 127.3, 126.1, 125.8, 121.9, 117.0, 116.7, 112.0, 108.9, 21.5, 21.4. IR (ATR, cm−1): ṽ = 1603 (s),1498 (m), 820 (s), 771 (m), 748 (vs), 734 (s), 725 (s), 612 (s), 556 (s). MS (EI, 70 eV): m/z (%) = 474 (92, M+), 473 (100), 472 (7), 238 (6), 237 (28), 236 (10), 230 (21), 229 (13), 228 (7), 222 (7), 215 (7). HRMS (ESI-TOF): calculated for C33H23N4 ([M + H]+) 475.1923, found 475.1926.
- 5,13-bis(4-fluorophenyl)pyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5c)
![Molecules 29 02159 i006]()
- According to general procedure A, the title compound 5c was obtained as an orange solid in 38% yield (44 mg, 0.091 mmol). Rf 0.78 (CH2Cl2/EtOAc 15:1). Mp. 318–320 °C. 1H NMR (500 MHz, CDCl3) δ = 8.86 (s, 1H), 8.32 (d, J = 8.5 Hz, 1H), 8.27 (d, J = 4.3 Hz, 1H), 7.89–7.84 (m, 3H), 7.68–7.65 (m, 1H), 7.54 (s, 1H), 7.50 (ddd, J = 8.3 Hz, J = 6.6 Hz, J = 1.2 Hz, 1H), 7.38 (d, J = 4.3 Hz, 1H), 7.36–7.28 (m, 6H). 13C NMR (126 MHz, CDCl3) δ = 163.5 (d, J = 250.0 Hz), 162.6 (d, J = 246.3 Hz), 153.4, 149.5, 149.3, 146.8, 146.7, 144.4, 139.9, 134.3 (d, J = 3.6 Hz), 133.3 (d, J = 3.2 Hz), 131.7, 130.4 (d, J = 8.7 Hz), 130.4 (d, J = 8.6 Hz),129.0, 127.6, 127.5, 127.2, 126.3, 126.3, 122.1, 117.5, 116.9, 116.2 (d, J = 21.8 Hz), 115.3 (d, J = 21.6 Hz), 112.4, 109.0. 19F NMR (471 MHz, CDCl3) δ = −111.4, −114.7. IR (ATR, cm−1): ṽ = 1603 (vs),1502 (vs), 1228 (vs), 1158 (s), 835 (vs), 802 (s), 769 (s), 748 (s), 736 (s), 610 (s), 563 (vs). MS (EI, 70 eV): m/z (%) = 482 (67, M+), 481 (100), 480 (42), 453 (13), 241 (27), 240 (71), 231 (16), 226 (37), 225 (32), 217 (16), 216 (17), 213 (14). HRMS (ESI-TOF): calculated for C31H17F2N4 ([M + H]+) 483.1421, found 483.1425.
- 5,13-bis(4-methoxyphenyl)pyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5e)
![Molecules 29 02159 i007]()
- According to general procedure A, the title compound 5e was obtained as an orange solid in 14% yield (16 mg, 0.032 mmol). Rf 0.85 (CH2Cl2/EtOAc 15:1). Mp. 298–301 °C. 1H NMR (250 MHz, CDCl3/TFA) δ = 9.05 (d, J = 4.9 Hz, 1H), 8.92 (s, 1H), 8.61 (d, J = 8.6 Hz, 1H), 8.44 (ddd, J = 8.6 Hz, J = 6.9 Hz, J = 1.4 Hz, 1H), 8.31–8.18 (m, 3H), 8.03–7.90 (m, 3H), 7.39–7.21 (m, 6H), 4.06 (s, 3H), 4.01 (s, 3H). 13C NMR (63 MHz, CDCl3/TFA) δ = 166.0, 163.3, 161.4, 148.6, 146.7, 145.3, 140.6, 140.3, 138.4, 136.1, 131.4, 131.3, 130.8, 130.1, 129.6, 128.1, 127.0, 126.6, 119.9, 119.7, 119.4, 118.4, 116.0, 115.9, 115.1, 111.1, 55.8, 55.7. IR (ATR, cm−1): ṽ = 1601 (s),1498 (s), 1243 (vs), 1175 (vs), 1158 (s), 1024 (vs), 835 (s), 785 (s), 762 (vs), 736 (s), 573 (s), 558 (vs). MS (EI, 70 eV): m/z (%) = 506 (100, M+), 505 (99), 463 (5) 462 (13), 420 (4), 419 (8), 254 (7), 253 (19), 232 (8), 231 (5), 210 (9), 197 (5), 196 (5). HRMS (ESI-TOF): calculated for C33H23N4O2 ([M + H]+) 507.1821, found 507.1828.
- 11-methyl-5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5g)
![Molecules 29 02159 i008]()
- According to general procedure A, the title compound 5g was obtained as an orange solid in 48% yield (59 mg, 0.128 mmol). Rf 0.75 (CH2Cl2/EtOAc 15:1).Mp. 319–322 °C. 1H NMR (500 MHz, CDCl3) δ = 8.80 (s, 1H), 8.17 (d, J = 4.3 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 7.87–7.84 (m, 2H), 7.64–7.54 (m, 7H), 7.53 (s, 1H), 7.40–7.36 (m, 4H), 2.44 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 153.3, 149.2, 148.2, 146.7, 143.7, 140.7, 138.8, 137.3, 136.1, 134.0, 129.4, 129.0, 128.7, 128.6, 128.6, 128.1, 127.4, 127.3, 127.1, 126.2, 126.2, 122.0, 117.2, 116.7, 111.8, 109.0, 22.0. IR (ATR, cm−1): ṽ = 1607 (s),1554 (m), 1492 (m), 1449 (m), 1403 (m), 1325 (m), 814 (m), 777 (vs), 754 (m), 703 (vs), 596 (m), 556 (s). MS (EI, 70 eV): m/z (%) = 460 (91, M+), 459 (100), 458 (11), 457 (7), 230 (28), 229 (29), 228 (15), 222 (18), 215 (8), 214 (13), 208 (7). HRMS (ESI-TOF): calculated for C32H21N4 ([M + H]+) 461.1766, found 461.1771.
- 9-methyl-5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5h)
![Molecules 29 02159 i009]()
- According to general procedure A, the title compound 5h was obtained as an orange solid in 38% yield (34 mg, 0.074 mmol). Rf 0.80 (CH2Cl2/EtOAc 15:1). Mp. 256–259 °C. 1H NMR (300 MHz, CDCl3) δ = 8.83 (s, 1H), 8.26 (d, J = 4.3 Hz, 1H), 7.92–7.85 (m, 2H), 7.69 (ddd, J = 6.8 Hz, J = 1.5 Hz, J = 1.0 Hz, 1H), 7.65–7.55 (m, 7H), 7.50 (ddd, J = 8.7 Hz, J = 1.5 Hz, J = 0.7 Hz, 1H), 7.43 (d, J = 4.3 Hz, 1H), 7.38–7.32 (m, 3H), 3.0 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 153.4, 150.1, 148.8, 146.9, 146.8, 143.4, 140.9, 139.1, 137.4, 136.9, 131.3, 129.4, 129.1, 128.7, 128.7, 128.1, 127.4, 127.1, 126.9, 125.8, 125.8, 122.2, 117.2, 116.4, 111.8, 109.0, 18.2. IR (ATR, cm−1): ṽ = 1601 (m),1552 (m), 1447 (m), 1428 (s), 1339 (m), 1323 (m), 781 (s), 762 (vs), 701 (vs), 604 (m), 567 (m), 556 (s). MS (EI, 70 eV): m/z (%) = 460 (81, M+), 459 (100), 458 (6), 457 (6), 445 (7), 230 (16), 229 (18), 228 (8), 222 (10), 214 (8). HRMS (ESI-TOF): calculated for C32H21N4 ([M + H]+) 461.1766, found 461.1775.
- 10,12-dimethyl-5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5i)
![Molecules 29 02159 i010]()
- According to general procedure A, the title compound 5i was obtained as an orange solid in 26% yield (34 mg, 0.072 mmol). Rf 0.82 (CH2Cl2/EtOAc 15:1). Mp. 317–320 °C. 1H NMR (500 MHz, CDCl3/TFA) δ = 9.12 (d, J = 4.9 Hz, 1H), 8.84 (s, 1H), 8.36 (s, 1H), 8.25 (s, 1H), 8.16 (d, J = 4.9 Hz, 1H), 7.95–7.87 (m, 2H), 7.76–7.64 (m, 6H), 7.58 (s, 1H), 7.40–7.35 (m, 2H), 2.77 (s, 3H), 2.15 (s, 3H). 13C NMR (126 MHz, CDCl3/TFA) δ = 165.2, 154.1, 147.5, 147.4, 145.6, 142.6, 142.0, 138.9, 137.5, 136.7, 135.2, 134.2, 132.1, 130.9, 130.4, 130.0, 129.4, 129.3, 127.1, 125.2, 121.3, 119.5, 119.2, 118.1, 117.3, 115.7, 112.8, 25.2, 22.8. IR (ATR, cm−1): ṽ = 1605 (s),1578 (m), 1550 (m), 1504 (m), 1451 (s), 1325 (m), 857 (m), 777 (s), 725 (s), 703 (vs), 558 (m). MS (EI, 70 eV): m/z (%) = 474 (89, M+), 473 (100), 471 (5), 470 (5), 399 (7), 237 (10), 236 (6), 229 (15), 228 (12), 214 (6). HRMS (ESI-TOF): calculated for C33H22N4 ([M + H]+) 475.1923, found 475.1934.
- 11-fluoro-5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5j)
![Molecules 29 02159 i011]()
- According to general procedure A, the title compound 5j was obtained as an orange solid in 55% yield (69 mg, 0.148 mmol). Rf 0.79 (CH2Cl2/EtOAc 15:1). Mp. 348–350 °C. 1H NMR (500 MHz, CDCl3) δ = 8.85 (s, 1H), 8.32 (dd, J = 9.3 Hz, J = 5.5 Hz, 1H), 8.24 (d, J = 4.3 Hz, 1H), 7.91–7.86 (m, 2H), 7.67–7.55 (m, 8H), 7.44 (d, J = 4.3 Hz, 1H), 7.38–7.35 (m, 2H), 7.26 (dd, J = 10.3 Hz, J = 2.8 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ = 160.1 (d, J = 248.7 Hz), 153.5, 149.5, 149.5, 147.0, 146.8, 146.4, 144.1 (d, J = 1.9 Hz), 141.1, 138.3, 137.3, 131.5 (d, J = 8.9 Hz), 129.5, 129.1, 128.7, 128.5, 128.4, 127.9 (d, J = 9.5 Hz), 127.8, 127.6, 126.2, 122.4 (d, J = 26.2 Hz), 117.5, 117.2, 112.0, 110.7 (d, J = 23.6 Hz), 109.2. 19F NMR (471 MHz, CDCl3) δ = −111.7. IR (ATR, cm−1): ṽ = 1488 (s),1175 (s), 830 (s), 777 (vs), 725 (s), 705 (vs), 593 (s), 552 (s), 474 (s), 460 (s), 443 (s). MS (EI, 70 eV): m/z (%) = 464 (89, M+), 463 (100), 462 (25), 435 (12), 232 (41), 231 (42), 230 (10), 218 (11), 217 (19). HRMS (ESI-TOF): calculated for C31H18FN4 ([M + H]+) 465.1516, found 465.1524.
- N,N-dimethyl-5,13-diphenylpyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinolin-11-amine (5k)
![Molecules 29 02159 i012]()
- According to general procedure A, the title compound 5k was obtained as an orange solid in 8% yield (11 mg, 0.021 mmol). Rf 0.82 (CH2Cl2/EtOAc 15:1). Mp. 130–134 °C. 1H NMR (500 MHz, CDCl3) δ = 8.81 (s, 1H), 8.25 (s, 2H), 7.91–7.88 (m, 2H), 7.62–7.57 (m, 5H), 7.57–7.55 (m, 3H), 7.45 (d, J = 4.3 Hz, 1H), 7.41–7.35 (m, 2H), 6.55 (d, J = 2.8 Hz, 1H), 2.96 (s, 6H). 13C NMR (126 MHz, CDCl3/TFA) δ = 148.3, 146.8, 146.7, 145.8, 138.9, 137.4, 134.9, 134.2, 134.0, 132.2, 130.8, 130.3, 130.1, 130.0, 129.8, 129.6, 129.3, 126.8, 122.4, 121.5, 120.4, 118.8, 117.8, 116.6, 112.0, 42.9. IR (ATR, cm−1): ṽ = 2920 (s),1607 (s), 1492 (s), 1449 (s), 1323 (s), 1123 (s), 775 (s), 699 (vs), 591 (s), 554 (s).MS (EI, 70 eV): m/z (%) = 489 (40, M+), 488 (24), 207 (14), 57 (25), 55 (20), 44 (100), 43 (28), 41 (23). HRMS (ESI-TOF): calculated for C33H24N5 ([M + H]+) 490.3032, found 490.2031.
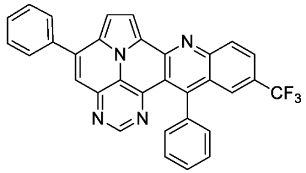
- 5,13-diphenyl-11-(trifluoromethyl)pyrimido[4′,5′,6′:9,1]pyrrolo[2′,1′,5′:4,5,6]quinolizino[3,2-b]quinoline (5l)
![Molecules 29 02159 i013]()
- According to general procedure A, the title compound 5l was obtained as an orange solid in 28% yield (38 mg, 0.075 mmol). Rf 0.85 (CH2Cl2/EtOAc 15:1). Mp. 311–313 °C. 1H NMR (500 MHz, CDCl3/TFA) δ = 9.10 (d, J = 4.9 Hz, 1H), 8.96 (s, 1H), 8.77 (d, J = 8.9 Hz, 1H), 8.56 (dd, J = 9.1 Hz, J = 1.8 Hz, 1H), 8.37 (s, 1H), 8.31 (s, 1H), 8.28 (d, J = 4.9 Hz, 1H), 7.95–7.89 (m, 2H), 7.86–7.82 (m, 1H), 7.81–7.71 (m, 5H), 7.41–7.37 (m, 2H). 13C NMR (126 MHz, CDCl3/TFA) δ = 167.4, 149.3, 147.4, 145.8, 141.4, 138.6, 137.3, 135.9 (q, J = 3.1 Hz), 133.8, 133.8, 132.7, 132.2, 132.1 (q, J = 34.7 Hz), 131.2, 130.3, 129.8, 129.4, 128.2 (q, J = 4.1 Hz), 127.1, 127.0, 122.4 (q, J = 273.1 Hz), 121.9, 121.5, 120.6, 119.7, 118.8, 116.9, 113.0. 19F NMR (471 MHz, CDCl3/TFA) δ = −63.8. IR (ATR, cm−1): ṽ = 1609 (m), 1311 (s), 1298 (s), 1117 (vs), 1067 (s), 985 (m), 837 (s), 779 (s), 701 (vs), 589 (s), 556 (s). MS (EI, 70 eV): m/z (%) = 514 (86, M+), 513 (100), 512 (5), 485 (5), 445 (4), 257 (11), 256 (14), 223 (4), 222 (13), 221 (4), 208 (5), 207 (4). HRMS (ESI-TOF): calculated for C32H18F3N4 ([M + H]+) 515.1483, found 515.1495.









3.2.2. General Procedure B for the Synthesis of Aryl(8-arylpyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6a–f)
- Phenyl(8-phenylpyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6a)
![Molecules 29 02159 i014]()
- According to general procedure B, the title compound 6a was obtained as a yellow solid in 68% yield (68 mg, 0.182 mmol). Rf 0.48 (heptane/EtOAc 1:2). Mp. 266–269 °C. 1H NMR (250 MHz, CDCl3) δ = 9.14 (s, 1H), 8.52 (s, 1H), 7.99–7.94 (m, 2H), 7.93 (s, 1H), 7.90–7.84 (m, 2H), 7.75–7.69 (m, 2H), 7.67–7.57 (m, 4H), 7.52–7.44 (m, 2H). 13C NMR (63 MHz, CDCl3) δ = 194.4, 155.1, 147.2, 144.9, 140.6, 137.2, 136.9, 133.5, 130.1, 129.6, 129.2, 128.9, 128.6, 128.1, 127.7, 126.3, 126.3, 124.7, 119.8, 113.9, 113.2. IR (ATR, cm−1): ṽ = 1644 (m), 1609 (s), 1455 (s), 1261 (s), 1238 (s), 1045 (s), 795 (s), 779 (s), 684 (vs), 637 (vs), 563 (vs). MS (EI, 70 eV): m/z (%) = 373 (16, M+), 372 (29), 346 (7), 345 (28), 344 (100), 316 (5), 240 (10), 187 (16), 172 (8). HRMS (ESI-TOF): calculated for C25H15N3O ([M + H]+) 374.1293, found 374.1294.
- p-tolyl(8-(p-tolyl)pyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6b)
![Molecules 29 02159 i015]()
- According to general procedure B, the title compound 6b was obtained as a yellow solid in 54% yield (54 mg, 0.135 mmol). Rf 0.52 (heptane/EtOAc 1:2). Mp. 258–261 °C. 1H NMR (300 MHz, CDCl3) δ = 9.12 (s, 1H), 8.47 (s, 1H), 7.88 (s, 1H), 7.86 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.1 Hz, 2H), 7.73–7.66 (m, 2H), 7.45–7.39 (m, 2H), 7.30–7.21 (m, 2H), 2.50 (s, 3H), 2.43 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 194.0, 155.1, 147.2, 144.8, 144.5, 140.5, 139.8, 134.6, 134.0, 130.3, 129.9, 129.2, 128.8, 128.4, 127.6, 126.2, 125.9, 124.6, 119.4, 113.6, 113.1, 21.7, 21.3. IR (ATR, cm−1): ṽ = 1607 (s), 1457 (s), 1344 (vs), 1267 (s), 1251 (s), 1043 (s), 903 (s), 824 (s), 777 (vs), 756 (s), 723 (s), 563 (vs). MS (EI, 70 eV): m/z (%) = 401 (19, M+), 400 (28), 373 (31), 372 (100), 371 (9), 201 (12), 186 (8), 179 (6), 178 (11), 91 (7). HRMS (ESI-TOF): calculated for C27H19N3O ([M + H]+) 402.1606, found 402.1602.
- (4-fluorophenyl)(8-(4-fluorophenyl)pyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6c)
![Molecules 29 02159 i016]()
- According to general procedure B, the title compound 6c was obtained as a yellow solid in 71% yield (71 mg, 0.173 mmol). Rf 0.48 (heptane/EtOAc 1:2). Mp. 317–320 °C. 1H NMR (500 MHz, CDCl3/TFA) δ = 9.25 (s, 1H), 9.17 (s, 1H), 8.63 (s, 1H), 8.41 (d, J = 5.0 Hz, 1H), 8.35 (d, J = 5.0 Hz, 1H), 8.00–7.87 (m, 4H), 7.44 (pt, J = 8.3 Hz, 2H), 7.30 (pt, J = 8.3 Hz, 2H). 13C NMR (126 MHz, CDCl3/TFA) δ = 193.9, 166.6 (d, J = 258.6 Hz), 164.9 (d, J = 254.6 Hz), 146.3, 144.6, 143.6, 136.7, 132.8 (d, J = 9.7 Hz), 131.9 (d, J = 8.9 Hz), 131.7, 130.6, 130.4, 130.3, 129.9, 122.9, 121.9, 121.2, 118.2, 117.5 (d, J = 22.1 Hz), 116.7 (d, J = 22.3 Hz). 19F NMR (471 MHz, CDCl3/TFA) δ = −101.5, −107.3. IR (ATR, cm−1): ṽ = 1609 (s), 1599 (s), 1589 (s), 1508 (s), 1453 (s), 1267 (s), 1243 (vs), 1232 (s), 1158 (s), 847 (s), 837 (s), 783 (vs), 563 (s). MS (EI, 70 eV): m/z (%) = 409 (14, M+), 408 (17), 382 (4), 381 (27), 380 (100), 379 (7), 259 (4), 258 (10), 205 (7), 190 (4), 95 (9). HRMS (ESI-TOF): calculated for C25H14F2N3O ([M + H]+) 410.1105, found 410.1113.
- (4-(dimethylamino)phenyl)(8-(4-(dimethylamino)phenyl)pyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6d)
![Molecules 29 02159 i017]()
- According to general procedure B, the title compound 6d was obtained as a yellow solid in 54% yield (54 mg, 0.118 mmol). Rf 0.29 (EtOAc). Mp. 255–258 °C. 1H NMR (500 MHz, CDCl3) δ = 9.11 (s, 1H), 8.39 (s, 1H), 7.91–7.75 (m, 6H), 7.63 (d, J = 4.6 Hz, 1H), 6.94–6.88 (m, 2H), 6.67–6.61 (m, 2H), 3.09 (s, 6H), 3.07 (s, 6H). 13C NMR (126 MHz, CDCl3) δ = 192.2, 155.0, 153.9, 151.2, 147.2, 144.6, 140.8, 132.7, 130.0, 129.7, 127.4, 126.4, 124.8, 124.7, 124.3, 124.3, 117.5, 113.1, 112.8, 112.4, 110.6, 40.3, 40.0. IR (ATR, cm−1): ṽ = 1597 (vs), 1523 (s), 1346 (s), 1284 (s), 1271 (s), 1261 (s), 1189 (s), 1179 (s), 1168 (s), 818 (s), 779 (s), 771 (s), 560 (s). MS (EI, 70 eV): m/z (%) = 459 (53, M+), 458 (39), 445 (22), 444 (28), 431 (34), 430 (100), 416 (35), 414 (16), 230 (18), 215 (13). HRMS (ESI-TOF): calculated for C29H26N5O ([M + H]+) 460.2137, found 460.2148.
- (4-methoxyphenyl)(8-(4-methoxyphenyl)pyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6e)
![Molecules 29 02159 i018]()
- According to general procedure B, the title compound 6e was obtained as a yellow solid in 51% yield (51 mg, 0.118 mmol). Rf 0.29 (heptane/EtOAc 1:3). Mp. 265–267 °C. 1H NMR (300 MHz, CDCl3) δ = 9.27 (s, 1H), 9.14 (s, 1H), 8.64 (s, 1H), 8.46–8.40 (m, 2H), 7.96–7.84 (m, 4H), 7.26 (d, J = 8.9 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 3.99 (s, 3H), 3.95 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 194.3, 165.4, 162.7, 146.3, 145.8, 143.0, 135.7, 133.1, 131.8, 131.7, 130.5, 130.1, 127.8, 126.6, 123.1, 122.9, 122.0, 121.1, 118.1, 115.8, 114.9, 55.8, 55.7. IR (ATR, cm−1): ṽ = 1597 (s), 1455 (s), 1253 (vs), 1166 (vs), 1041 (s), 1020 (s), 832 (s), 775 (s), 604 (s), 575 (s), 560 (s), 536 (s). MS (EI, 70 eV): m/z (%) = 433 (16, M+), 432 (20), 405 (26), 404 (100), 389 (11), 361 (13), 217 (26), 180 (9), 265 (7). HRMS (ESI-TOF): calculated for C27H20N3O3 ([M + H]+) 434.1505, found 434.1518.
- (4-(trifluoromethyl)phenyl)(8-(4-(trifluoromethyl)phenyl)pyrimido[4,5,6-ij]pyrrolo[2,1,5-de]quinolizin-4-yl)methanone (6f)
![Molecules 29 02159 i019]()
- According to general procedure B, the title compound 6f was obtained as a yellow solid in 62% yield (62 mg, 0.121 mmol). Rf 0.52 (heptane/EtOAc 1:2). Mp. 309–311 °C. 1H NMR (500 MHz, CDCl3/TFA) δ = 9.23 (s, 1H), 9.18 (s, 1H), 8.66 (s, 1H), 8.41 (d, J = 5.0 Hz, 1H), 8.30 (d, J = 5.0 Hz, 1H), 8.04 (d, J = 8.1 Hz, 2H), 8.02–7.94 (m, 4H), 7.85 (d, J = 7.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ = 194.3, 146.2, 144.1, 143.8, 138.8, 137.6, 137.0, 135.3 (q, J = 33.0 Hz), 133.4 (q, J = 33.3 Hz), 131.7, 130.9, 130.2, 130.0, 129.9, 126.9 (q, J = 3.8 Hz), 126.2 (q, J = 3.8 Hz), 123.6, 123.5 (q, J = 272.7 Hz), 123.3, 123.3 (q, J = 272.9 Hz), 122.6, 120.8, 117.8. 19F NMR (471 MHz, CDCl3) δ = −63.2, −63.5. IR (ATR, cm−1): ṽ = 1325 (vs), 1166 (s), 1109 (vs), 1067 (s), 1057 (s), 1049 (s), 1018 (s), 845 (m), 787 (s), 670 (m), 563 (m). MS (EI, 70 eV): m/z (%) = 509 (12, M+), 508 (20), 482 (5), 481 (28), 480 (100), 255 (7), 240 (8), 145 (9). HRMS (ESI-TOF): calculated for C27H14F6N3O ([M + H]+) 510.1041, found 510.1055.






Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Delcamp, J.H.; Yella, A.; Holcombe, T.W.; Nazeeruddin, M.K.; Grätzel, M. The molecular engineering of organic sensitizers for solar-cell applications. Angew. Chem. Int. Ed. 2013, 52, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Balli, H.; Zeller, M. Neue Heteroarene: Synthese und spektrale Daten von Indolizino[6,5,4,3-aij ]chinolin («Ullazin») und einigen Derivaten. Helv. Chim. Acta 1983, 66, 2135–2139. [Google Scholar] [CrossRef]
- Feng, J.; Jiao, Y.; Ma, W.; Nazeeruddin, M.K.; Grätzel, M.; Meng, S. First Principles Design of Dye Molecules with Ullazine Donor for Dye Sensitized Solar Cells. J. Phys. Chem. C 2013, 117, 3772–3778. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheema, H.; McNamara, L.; Hunt, L.A.; Hammer, N.I.; Delcamp, J.H. Ullazine Donor-π bridge-Acceptor Organic Dyes for Dye-Sensitized Solar Cells. Chem. Eur. J. 2018, 24, 5939–5949. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C. Ullazine-based materials: Towards novel opportunities in organic electronics. J. Mater. Chem. C 2018, 6, 11943–11950. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Xu, Y. Theoretical screening of high-efficiency sensitizers with D-π-A framework for DSSCs by altering promising donor group. Sol. Energy. 2020, 196, 146–156. [Google Scholar] [CrossRef]
- Le Bao, Q.; Thogiti, S.; Koyyada, G.; Kim, J.H. Synthesis and photovoltaic performance of novel ullazine-based organic dyes for dye-sensitized solar cells. Jpn. J. Appl. Phys. 2019, 58, 12011. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, I.; König, B. Synthesis of pyrrolo1,2-aquinolines and ullazines by visible light mediated one- and twofold annulation of N-arylpyrroles with arylalkynes. Chem. Commun. 2016, 52, 8695–8698. [Google Scholar] [CrossRef] [PubMed]
- Pierrat, P.; Hesse, S.; Cebrián, C.; Gros, P.C. Controlling charge-transfer properties through a microwave-assisted mono- or bis-annulation of dialkynyl-N-(het)arylpyrroles. Org. Biomol. Chem. 2017, 15, 8568–8575. [Google Scholar] [CrossRef]
- Kanno, K.; Liu, Y.; Iesato, A.; Nakajima, K.; Takahashi, T. Chromium-mediated synthesis of polycyclic aromatic compounds from halobiaryls. Org. Lett. 2005, 7, 5453–5456. [Google Scholar] [CrossRef]
- Drigo, N.A.; Paek, S.; Huckaba, A.J.; Schouwink, P.A.; Tabet, N.; Nazeeruddin, M.K. Approaches for Selective Synthesis of Ullazine Donor-Acceptor Systems. Chem. Eur. J. 2017, 23, 17209–17212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Gautam, P.; Chan, J.M.W. Symmetrical and unsymmetrical fluorine-rich ullazines via controlled cycloaromatizations. Org. Chem. Front. 2020, 7, 787–795. [Google Scholar] [CrossRef]
- Qiao, H.; Deng, Y.; Peng, R.; Wang, G.; Yuan, J.; Tan, S. Effect of π-spacers and anchoring groups on the photovoltaic performances of ullazine-based dyes. RSC Adv. 2016, 6, 70046–70055. [Google Scholar] [CrossRef]
- Wan, D.; Li, X.; Jiang, R.; Feng, B.; Lan, J.; Wang, R.; You, J. Palladium-Catalyzed Annulation of Internal Alkynes: Direct Access to π-Conjugated Ullazines. Org. Lett. 2016, 18, 2876–2879. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Fu, Y.; Dmitrieva, E.; Weigand, J.J.; Popov, A.; Berger, R.; Liu, J.; Feng, X. Polycyclic Aromatic Hydrocarbons Containing A Pyrrolopyridazine Core. ChemPlusChem 2019, 84, 613–618. [Google Scholar] [CrossRef]
- Liu, J.; Feng, X. Bottom-Up Synthesis of Nitrogen-Doped Polycyclic Aromatic Hydrocarbons. Synlett 2020, 31, 211–222. [Google Scholar] [CrossRef]
- Berger, R.; Wagner, M.; Feng, X.; Müllen, K. Polycyclic aromatic azomethine ylides: A unique entry to extended polycyclic heteroaromatics. Chem. Sci. 2015, 6, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Tokimaru, Y.; Nozaki, K. Isoquinolino4,3,2-dephenanthridine: Synthesis and its use in 1,3-dipolar cycloadditions to form nitrogen-containing polyaromatic hydrocarbons. Chem. Commun. 2015, 51, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Tokimaru, Y.; Ito, S.; Nozaki, K. Synthesis of Pyrrole-Fused Corannulenes: 1,3-Dipolar Cycloaddition of Azomethine Ylides to Corannulene. Angew. Chem. Int. Ed. 2017, 56, 15560–15564. [Google Scholar] [CrossRef]
- Hager, J.; Kang, S.; Chmielewski, P.J.; Lis, T.; Kim, D.; Stępień, M. Acenaphthylene-fused ullazines: Fluorescent π-extended monopyrroles with tunable electronic gaps. Org. Chem. Front. 2022, 9, 3179–3185. [Google Scholar] [CrossRef]
- Miao, D.; Aumaitre, C.; Morin, J.-F. Photochemical synthesis of π-extended ullazine derivatives as new electron donors for efficient conjugated D–A polymers. J. Mater. Chem. C 2019, 7, 3015–3024. [Google Scholar] [CrossRef]
- Hou, D.; Balli, H. A Novel Heterocyclic Ring System: Synthesis and Spectral Data of 4,8,9b-Triazacyclopenta[c,d]phenalene. Helv. Chim. Acta 1992, 75, 2608–2612. [Google Scholar] [CrossRef]
- Janke, S.; Boldt, S.; Nakielski, P.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis and Properties of 5-Azaullazines. J. Org. Chem. 2023, 88, 10470–10482. [Google Scholar] [CrossRef] [PubMed]
- Boldt, S.; Parpart, S.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis and Properties of Aza-ullazines. Angew. Chem. Int. Ed. 2017, 56, 4575–4578. [Google Scholar] [CrossRef]
- Ge, Q.; Li, B.; Wang, B. Synthesis of substituted benzoijimidazo2,1,5-dequinolizine by rhodium(III)-catalyzed multiple C-H activation and annulations. Org. Biomol. Chem. 2016, 14, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.L.; Ellul, C.E.; Macgregor, S.A.; McMullin, C.L.; Singh, K. Experimental and DFT Studies Explain Solvent Control of C-H Activation and Product Selectivity in the Rh(III)-Catalyzed Formation of Neutral and Cationic Heterocycles. J. Am. Chem. Soc. 2015, 137, 9659–9669. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Y.; Sun, Z.; Zhang, J.; Liu, M.; Zhang, C.; Zhang, Q.; Wang, H.; Liu, X. Synthesis, Characterization, and Properties of Bis-BN Ullazines. Org. Lett. 2018, 20, 2806–2810. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, L.; Li, C.; Jin, M.; Zhang, Y.; Ye, J.; Chen, Y.; Wu, X.; Liu, X. BN/BO-Ullazines and Bis-BO-Ullazines: Effect of BO Doping on Aromaticity and Optoelectronic Properties. J. Org. Chem. 2021, 86, 12507–12516. [Google Scholar] [CrossRef] [PubMed]
- Polkaehn, J.; Molenda, R.; Cordero, M.A.; Lochbrunner, S.; Boldt, S.; Ehlers, P.; Villinger, A.; Langer, P. Synthesis and Properties of 5,7-Diazaullazines. J. Org. Chem. 2024, 89, 2169–2181. [Google Scholar] [CrossRef]
- Polkaehn, J.; Thom, R.; Ehlers, P.; Villinger, A.; Langer, P. π-Expanded azaullazines: Synthesis of quinolino-azaullazines by Povarov reaction and cycloisomerisation. Org. Biomol. Chem. 2024. [Google Scholar] [CrossRef]
- Spruner von Mertz, F.; Molenda, R.; Boldt, S.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis and Properties of Diphenylbenzojnaphtho2,1,8-def2,7phenanthrolines. Chem. Eur. J. 2023, 29, e202204011. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, J.; Xie, L.; Fan, H.; Sheng, X.; Du, Y.; Liu, G.; Hu, H.; Jiang, Y.; Chen, M. Iron-catalyzed divergent approach to naphthyridinones and quinolinones: Leveraging Povarov and carbonyl-alkyne metathesis reactions of electron deficient alkynes. Org. Chem. Front. 2023, 10, 5505–5511. [Google Scholar] [CrossRef]
- Chakraborty, B.; Kar, A.; Chanda, R.; Jana, U. Application of the Povarov Reaction in Biaryls under Iron Catalysis for the General Synthesis of Dibenzoa, cAcridines. J. Org. Chem. 2020, 85, 9281–9289. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, M.; Frey, A.; Rettmann, A.; Thom, R.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis of Dibenzotropones by Alkyne-Carbonyl Metathesis. J. Org. Chem. 2021, 86, 14420–14432. [Google Scholar] [CrossRef]
- CCDC 2332651 Contains the Supplementary Crystallographic Data for This Paper. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 19 February 2024).
- Paenurk, E.; Gershoni-Poranne, R. Simple and efficient visualization of aromaticity: Bond currents calculated from NICS values. Phys. Chem. Chem. Phys. 2022, 24, 8631–8644. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef]
- Vardanyan, A.; Boldt, S.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis and Properties of 1-Azapyrenes. J. Org. Chem. 2022, 87, 11296–11308. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, A.; Argüello Cordero, M.A.; Lochbrunner, S.; Villinger, A.; Ehlers, P.; Langer, P. Synthesis and Properties of 4- and 10-Benzoyl-1-azapyrenes. J. Org. Chem. 2024, 89, 2155–2168. [Google Scholar] [CrossRef]
- Parpart, S.; Boldt, S.; Ehlers, P.; Langer, P. Synthesis of Unsymmetrical Aza-Ullazines by Intramolecular Alkynyl-Carbonyl Metathesis. Org. Lett. 2018, 20, 122–125. [Google Scholar] [CrossRef]
- Pommerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R.F.; Bässler, H.; Porsch, M.; Daub, J. Efficient two layer leds on a polymer blend basis. Adv. Mater. 1995, 7, 551–554. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Xiang, J.; Zheng, L.; Chen, F.; Dang, Q.; Bai, X. A cascade reaction consisting of Pictet-Spengler-type cyclization and Smiles rearrangement: Application to the synthesis of novel pyrrole-fused dihydropteridines. Org. Lett. 2007, 9, 765–767. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | eq. FeCl3 | Acid (eq.) | Solvent | T [°C] | t1 [h] | t2 [h] | Yield [a] [%] |
| 1 | 0.1 | p-TsOH∙H2O (30) | toluene | 100 | 3 | 2 | traces |
| 2 | 0.3 | p-TsOH∙H2O (30) | xylene | 140 | 3 | 2 | 29 |
| 3 | 0.6 | p-TsOH∙H2O (30) | xylene | 140 | 3 | 2 | 34 |
| 4 | 1 | p-TsOH∙H2O (30) | xylene | 140 | 3 | 2 | 38 |
| 5 | 4 | - | xylene | 140 | 3 | - | 21 |
| 6 | 1 | p-TsOH∙H2O (10) | xylene | 140 | 3 | 2 | 23 |
| 7 | 1 | p-TsOH∙H2O (20) | xylene | 140 | 3 | 2 | 27 |
| 8 | 1 | p-TsOH∙H2O (30) | xylene | 140 | 3 | 6 | 45 |
| 9 | 1 | p-TsOH∙H2O (30) | xylene | 140 | 3 | 16 | 45 |
| 10 | 1 | p-TsOH∙H2O (40) | xylene | 140 | 3 | 6 | 43 |
| 11 | 1 | MsOH (30) | xylene | 140 | 3 | 6 | 40 |
 | |||||
|---|---|---|---|---|---|
| Entry | Acid (eq.) | Solvent | T [°C] | t [h] | Yield [a] [%] |
| 1 | p-TsOH∙H2O (30) | - | 120 | 16 | 57 |
| 2 | p-TsOH∙H2O (30) | xylene | 120 | 16 | 68 |
| 3 | p-TsOH∙H2O (30) | xylene | 140 | 16 | 59 |
| 4 | p-TsOH∙H2O (20) | xylene | 120 | 16 | 67 |
| 5 | p-TsOH∙H2O (10) | xylene | 120 | 16 | 53 |
| 6 | MsOH (20) | xylene | 120 | 16 | 27 |
| 7 | p-TsOH∙H2O (20) | xylene | 120 | 6 | 68 |
 |  |  |  | |
|---|---|---|---|---|
| 5a | 6a | 7 [30] | 8 [29] | |
| λ1,abs [nm] | 497 | 420 | 471 | 422 |
| ελ1 [104 L·mol−1cm−1] | 0.8 | 2.0 | 1.1 | 1.7 |
| λ1,em [nm] | 530 | 462 | 515 | 439 |
| Φ | 0.52 | 0.10 | 0.26 | 0.35 |
| EOx. [V vs. Fc/Fc+] | 0.95 | 1.08 | 0.72 | 1.00 |
| EHOMO/CV [eV] | −5.65 | −5.76 | −5.41 | −5.69 |
| EHOMO/DFT [eV] | −5.53 | −5.73 | −5.22 | −5.60 |
| ELUMO/DFT [eV] | −2.43 | −2.04 | −2.09 | −1.90 |
| Egap [eV] | 3.10 | 3.69 | 3.13 | 3.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polkaehn, J.; Ehlers, P.; Villinger, A.; Langer, P. Divergent Synthesis of 5,7-Diazaullazines Derivatives through a Combination of Cycloisomerization with Povarov or Alkyne–Carbonyl Metathesis. Molecules 2024, 29, 2159. https://doi.org/10.3390/molecules29092159
Polkaehn J, Ehlers P, Villinger A, Langer P. Divergent Synthesis of 5,7-Diazaullazines Derivatives through a Combination of Cycloisomerization with Povarov or Alkyne–Carbonyl Metathesis. Molecules. 2024; 29(9):2159. https://doi.org/10.3390/molecules29092159
Chicago/Turabian StylePolkaehn, Jonas, Peter Ehlers, Alexander Villinger, and Peter Langer. 2024. "Divergent Synthesis of 5,7-Diazaullazines Derivatives through a Combination of Cycloisomerization with Povarov or Alkyne–Carbonyl Metathesis" Molecules 29, no. 9: 2159. https://doi.org/10.3390/molecules29092159