
DNA Methylation Analysis of Growth Differences between Upright and Inverted Cuttings of Populus yunnanensis
 ,
,
Abstract
:1. Introduction
2. Results
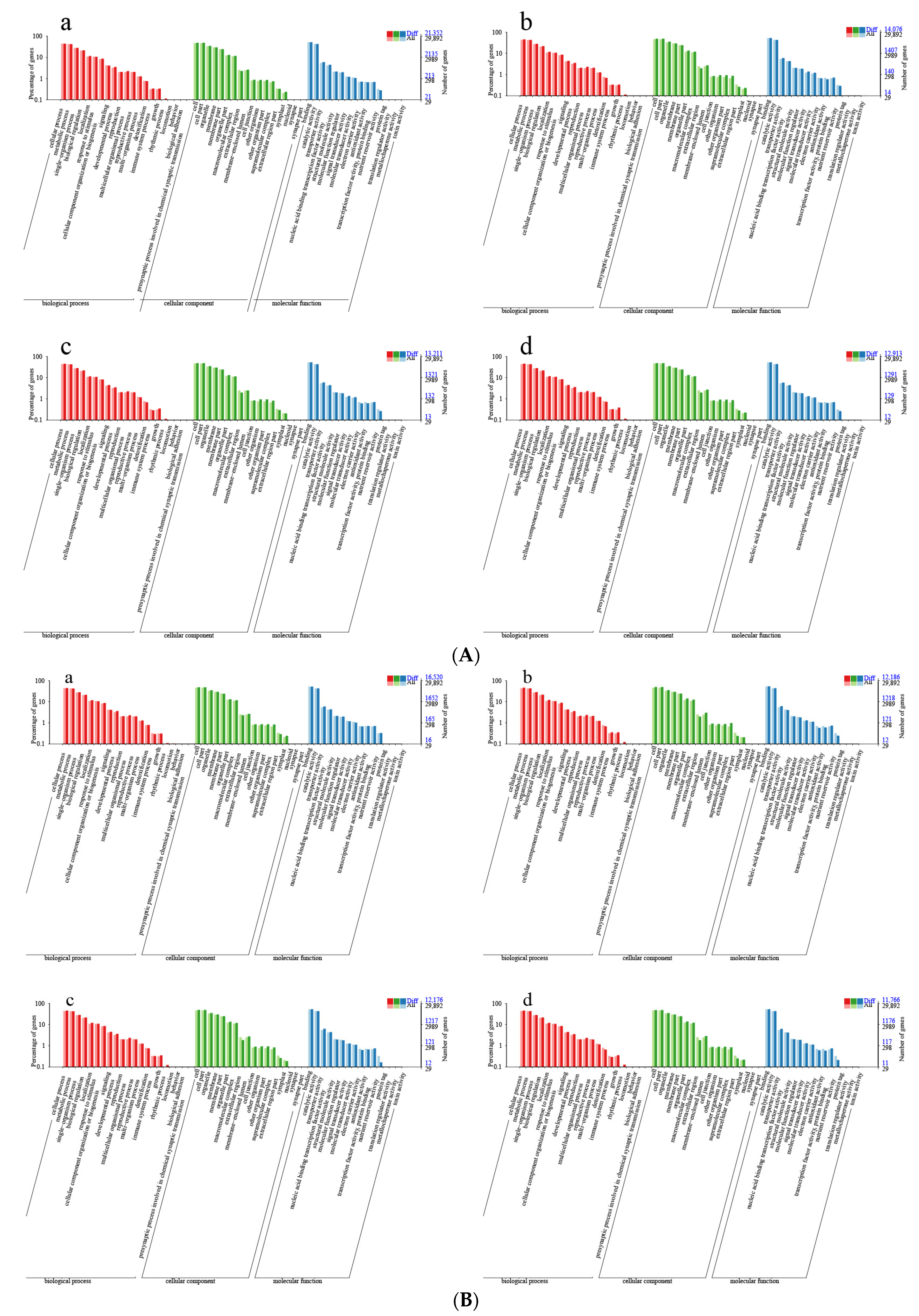
2.1. Methylation Profiles in P. yunnanensis Stem Tip
2.2. Methylation Changed in Different Growth Stages and Cutting Methods in P. yunnanensis
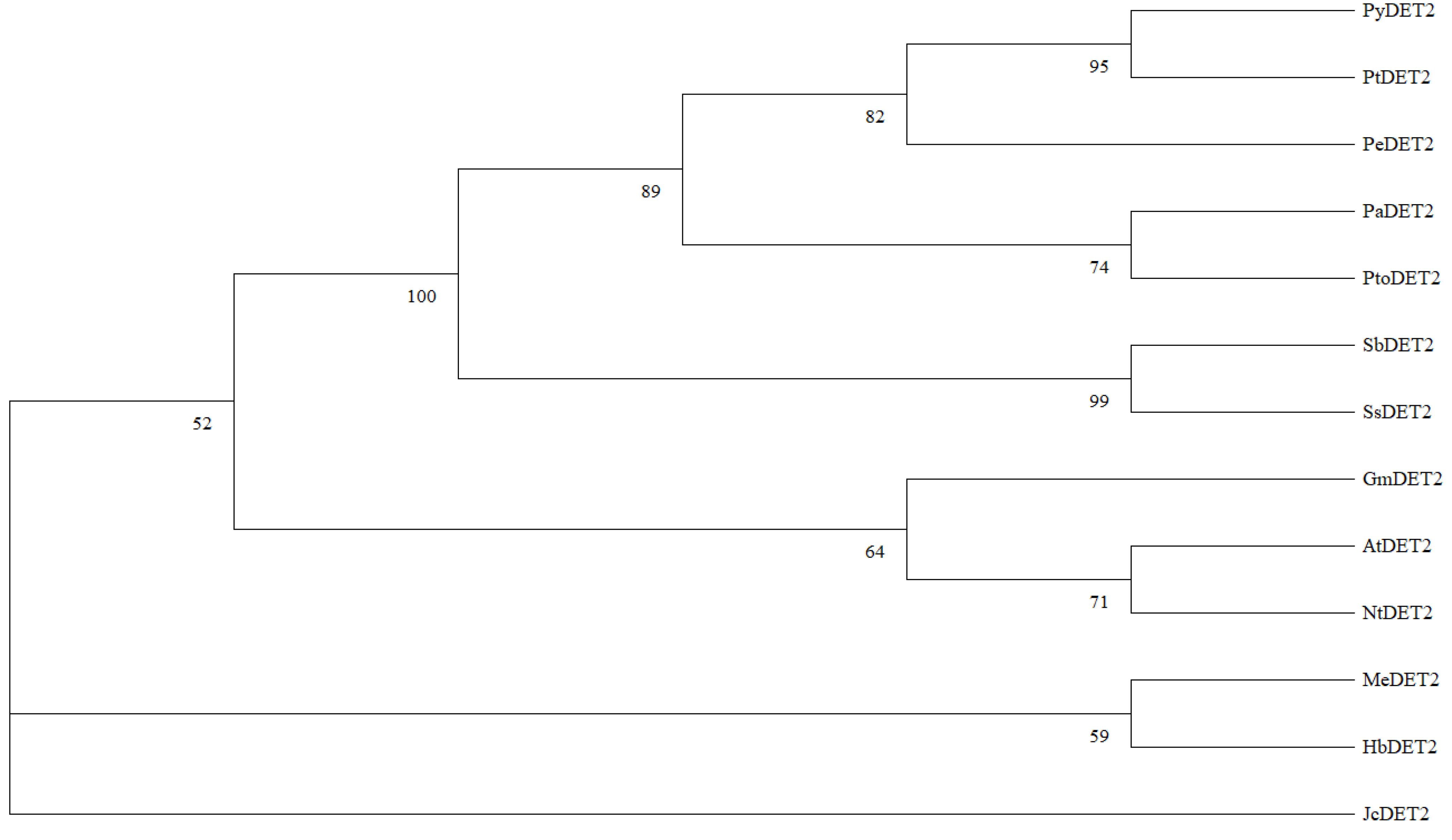
2.3. Sequence Analysis of PyDET2 Gene
3. Discussion
4. Materials and Methods
4.1. Plant Material Cultivation and Collection
4.2. Data Filtering and Methylation Level Analysis
4.3. Screening of Differential Methylation Regions
4.4. Bisulfite Sequencing of Differentially Methylated Genes
4.5. RNA Isolation and qPCR Analyses
4.6. Functional Analysis of PyDET2 Gene
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waddington, C.H. The Strategy of the Genes; Routledge: Oxford, UK, 2014. [Google Scholar]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Zhou, M.; Coruh, C.; Xu, G.; Martins, L.M.; Bourbousse, C.; Lambolez, A.; Law, J.A. The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. Nat. Commun. 2022, 13, 244. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolome, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, L.; Feng, Y.; Wei, Y.; Yue, X.; He, W.; Zhang, H.; An, L. Chilling- and Freezing- Induced Alterations in Cytosine Methylation and Its Association with the Cold Tolerance of an Alpine Subnival Plant, Chorispora bungeana. PLoS ONE 2015, 10, e0135485. [Google Scholar] [CrossRef] [PubMed]
- Popova, O.V.; Dinh, H.Q.; Aufsatz, W.; Jonak, C. The RdDM Pathway Is Required for Basal Heat Tolerance in Arabidopsis. Mol. Plant 2013, 6, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Liu, M.; Zhou, R.; Jiang, F.; Wu, Z. Relationship between melatonin and abscisic acid in response to salt stress of tomato. Sci. Hortic. 2021, 285, 110176. [Google Scholar] [CrossRef]
- Ahmad, N.; Naeem, M.; Ali, H.; Alabbosh, K.F.; Hussain, H.; Khan, I.; Siddiqui, S.A.; Khan, A.A.; Iqbal, B. From challenges to solutions: The impact of melatonin on abiotic stress synergies in horticultural plants via redox regulation and epigenetic signaling. Sci. Hortic. 2023, 321, 112369. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, X.; Liu, H.; Zhang, M.; Liao, W. DNA methylation in tomato fruit ripening. Physiol. Plant. 2022, 174, e13627. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Che, P.; Zhou, X.; Duan, A.; Wang, D.; Xin, P. A survey of research progress on gene resources of Populus yunnanensis. J. Southwest For. Univ. 2010, 30, 83–94. [Google Scholar]
- Zhou, A.-P.; Gan, P.-H.; Zong, D.; Fei, X.; Zhong, Y.-Y.; Li, S.-Q.; Yu, J.-D.; He, C.-Z. Bark tissue transcriptome analyses of inverted Populus yunnanensis cuttings reveal the crucial role of plant hormones in response to inversion. PeerJ 2019, 7, e7740. [Google Scholar] [CrossRef] [PubMed]
- Lukens, L.N.; Zhan, S. The plant genome’s methylation status and response to stress: Implications for plant improvement. Curr. Opin. Plant Biol. 2007, 10, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.-J.; Liang, L.-X.; Diao, S.; Su, X.-H.; Zhang, B.-Y. Genome-wide analysis of day/night DNA methylation differences in Populus nigra. PLoS ONE 2018, 13, e0190299. [Google Scholar] [CrossRef]
- Zhou, A.-P.; Zhong, Y.-Y.; Li, S.-Q.; Fei, X.; Gan, P.-H.; Zong, D.; He, C.-Z. Genome-wide identification of polar auxin transporter gene families reveals a possible new polar auxin flow in inverted cuttings of Populus yunnanensis. Gene 2021, 772, 145349. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Seymour, D.K.; Koenig, D.; Hagmann, J.; Becker, C.; Weigel, D. Evolution of DNA Methylation Patterns in the Brassicaceae is Driven by Differences in Genome Organization. PLoS Genet. 2014, 10, e1004785. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Yokota, T. Biosynthesis and metabolism of brassinosteroids. Annu. Rev. Plant Biol. 2003, 54, 137–164. [Google Scholar] [CrossRef]
- Park, C.H.; Jang, M.-S.; Yun, J.H.; Lee, J.E.; Kim, M.K.; Park, S.-C.; Kim, S.-K. Effects of secondary mutation in det2-1 on root growth and development in Arabidopsis. J. Plant Biol. 2014, 57, 255–263. [Google Scholar] [CrossRef]
- Zeng, H.; Tang, Q.; Hua, X. Arabidopsis Brassinosteroid Mutants det2-1 and bin2-1 Display Altered Salt Tolerance. J. Plant Growth Regul. 2010, 29, 44–52. [Google Scholar] [CrossRef]
- Qiao, Z.; Li, J.; Zhang, X.; Guo, H.; He, C.; Zong, D. Genome-Wide Identification, Expression Analysis, and Subcellular Localization of DET2 Gene Family in Populus yunnanensis. Genes 2024, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yang, C.; Wang, X. Brassinosteroids can regulate cellulose biosynthesis by controlling the expression of CESA genes in Arabidopsis. J. Exp. Bot. 2011, 62, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, Y.; Guo, Y.; Shou, H.; Du, J. PagDET2 promotes cambium cell division and xylem differentiation in poplar stem. Front. Plant Sci. 2022, 13, 923530. [Google Scholar] [CrossRef] [PubMed]
- Ze-hua, L.; Juan, D.; Xue-jiao, H.; Shu-tang, Z.; Ying-li, L.; Meng-zhu, L. Cloning and Characterization of Brassinosteriod Biosynthesis-related Gene DET2 in Poplar. For. Res. 2020, 33, 85–92. [Google Scholar]
- Deng, W.; Lv, L.; Luo, K.; Li, Y. Effect of Overexpression of Steroid 5α-Reductase Gene on Growth of Populus tomentosa Carr. Plant Physiol. Commun. 2008, 44, 399–403. [Google Scholar]
- Weige, H.; Bodi, L.; Jiebing, K.; Pingan, H.; Zhihao, X.; Jinxiang, W. Functional Characterization of the Steroid Reductase GenesGmDET2aandGmDET2bfromGlycine max. Int. J. Mol. Sci. 2018, 19, 726. [Google Scholar]
- Luo, X.-M.; Lin, W.-H.; Zhu, S.; Zhu, J.-Y.; Sun, Y.; Fan, X.-Y.; Cheng, M.; Hao, Y.; Oh, E.; Tian, M.; et al. Integration of Light- and Brassinosteroid-Signaling Pathways by a GATA Transcription Factor in Arabidopsis. Dev. Cell 2010, 19, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nagpal, P.; Vitart, V.; McMorris, T.C.; Chory, J. A role for brassinosteroids in light-dependent development of Arabidopsis. Science 1996, 272, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, M.; Nemeth, K.; Koncz-Kalman, Z.; Mathur, J.; Kauschmann, A.; Altmann, T.; Redei, G.P.; Nagy, F.; Schell, J.; Koncz, C. Brassinosteroids rescue the deficiency of CYP90, a cytochrome P450, controlling cell elongation and de-etiolation in Arabidopsis. Cell 1996, 85, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Workman, R.; Zuzar, P.C.; Davd, M.; Dursi, J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, M.; Quick, J.; Egan, R.; Eun, L.J.; Celniker, S.; Rk, N.; Loman, N.; La, P.; Brown, J. De Novoidentification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing; Cold Spring Harbor Laboratory: New York, NY, USA, 2016. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | SeqNum | MeanQual | Mapped | Unmapped | Ratio (%) |
|---|---|---|---|---|---|
| JD1 | 1,907,103 | 9.07 | 1,893,080 | 14,023 | 99.26 |
| JD2 | 2,263,391 | 9.00 | 2,248,376 | 15,015 | 99.34 |
| JD3 | 2,561,229 | 9.07 | 2,541,902 | 19,327 | 99.25 |
| JZ1 | 2,383,091 | 9.07 | 2,363,922 | 19,169 | 99.20 |
| JZ2 | 2,382,629 | 9.55 | 2,366,818 | 15,811 | 99.34 |
| JZ3 | 2,275,019 | 9.51 | 2,262,031 | 12,988 | 99.43 |
| SD1 | 2,462,841 | 9.54 | 2,431,027 | 31,814 | 98.71 |
| SD2 | 2,615,083 | 9.55 | 2,588,305 | 26,778 | 98.98 |
| SD3 | 2,090,749 | 9.00 | 2,056,227 | 34,522 | 98.35 |
| SZ1 | 3,101,285 | 9.08 | 3,054,641 | 46,644 | 98.50 |
| SZ2 | 2,414,738 | 8.92 | 2,372,104 | 42,634 | 98.23 |
| SZ3 | 2,246,146 | 9.06 | 2,216,803 | 29,343 | 98.69 |
| ID | CpG | CHG | CHH | 6mA |
|---|---|---|---|---|
| JD1 | 3,762,802 | 5,329,392 | 24,840,600 | 27,585,968 |
| JD2 | 4,737,441 | 7,551,680 | 36,421,035 | 43,349,990 |
| JD3 | 4,203,995 | 6,893,669 | 33,014,063 | 38,648,190 |
| JZ1 | 5,566,422 | 8,458,870 | 41,736,818 | 51,431,038 |
| JZ2 | 5,207,927 | 7,815,594 | 38,080,584 | 45,926,680 |
| JZ3 | 5,301,295 | 7,661,277 | 37,110,631 | 44,386,235 |
| SD1 | 4,332,887 | 6,400,612 | 30,245,830 | 34,609,545 |
| SD2 | 4,819,742 | 7,128,564 | 34,175,017 | 40,030,356 |
| SD3 | 2,566,496 | 4,121,957 | 18,423,471 | 19,866,134 |
| SZ1 | 5,556,780 | 9,377,372 | 46,555,303 | 60,528,769 |
| SZ2 | 2,992,772 | 5,201,162 | 23,789,123 | 26,780,834 |
| SZ3 | 3,634,195 | 5,618,731 | 26,098,363 | 30,000,662 |
| Element | Sequence (5′-3′) | Location | Number of Elements | Function |
|---|---|---|---|---|
| ABRE | ACGTG | 1875+ | 1 | Abscisic acid responsiveness cis-element |
| LTR | CCGAAA | 752−, 1172−, 1235−, 1522− | 4 | Low-temperature responsiveness cis-element |
| TC-rich repeats | ATTCTCTAAC | 1360+ | 1 | Defense and stress responsiveness cis-element |
| TCA-element | CCATCTTTTT | 1126−, 1753− | 2 | Salicylic acid responsiveness element |
| ARE | AAACCA | 601−, 669−, 1202+, 1207+, 1298+, 1675+, 1927 | 7 | Anaerobic induction regulatory cis-element |
| G-Box | CACGTT | 1874− | 1 | Light responsiveness cis-element |
| CAT-box | GCCACT | 412− | 1 | Meristem expression-related cis-element |
| CAAT-box | CCAAT | 667−, 834−, 889−, 1184+, 1187−, 1369+, 1378+, 1432−, 1462−, 1560+, 1724+ | 11 | Promoter and enhancer regions cis-element |
| TATA-box | TATA | 205+, 211−, 212+, 217−, 219+, 625−, 626−, 627−, 628+, 1436−, 1443+, 1444−, 1479−, 1489−, 1490−, 1705−, 1706−, 1707−, 1708−, 1743−, 1775−, 1776−, 1777−, 1778−, 1779+, 1780− | 26 | Core promoter element around −30 of transcription start |
| P-box | CCTTTTG | 1038+ | 1 | Gibberellin-responsive element |
| Box 4 | ATTAAT | 121+, 1215− | 2 | Part of a conserved DNA module involved in light responsiveness |
| AE-box | AGAAACTT | 1970− | 1 | Part of a module for light response |
| Sp1 | GGGCGG | 180− | 1 | Light-responsive element |
| GT1-motif | GGTTAA | 1815− | 1 | Light-responsive element |
| TCCC-motif | TCTCCCT | 92− | 1 | Part of a light-responsive element |
| GA-motif | ATAGATAA | 1393− | 1 | Part of a light-responsive element |
| TCT-motif | TCTTAC | 1501+ | 1 | Part of a light-responsive element |
| Name | Primer Sequence | Amplification Length | Site (to TSS) |
|---|---|---|---|
| PyMORG1 | F: GGTTTTGAGTTAAAATAAATAATTAAATTA | 191 | −534 |
| R: ACCAAAATTTCTCAAAAAATAAACAC | |||
| PySOD1 | F: TTAAAAGTTGTTATTATTAGATTGAAGGTT | 207 | −4055 |
| R: ACCACAATAATATTATCCCCTCTTC | |||
| PyUNC | F: AGGTGTTTTTTAAGAAGAGGTATTTATATT | 155 | +38,126 |
| R: T AAAAAATCATCCTTCCACTATACCTC | |||
| PyDET2 | F: TGTGGGTTATAATTAGGGGTGTTTA | 234 | −2069 |
| R: AATTAATATTTAATATCAAATTTTTCCTTC | |||
| PyMYBP | F: TTATTTGGGTTTGTATTTATATATGTGAGA | 254 | +22,128 |
| R: TCCTTTCAAAACCACTAAAAATTTAC | |||
| PyfrmA | F: ATTGGTTTAGTTGTTAGGATAAAGG | 111 | −2027 |
| Name | Primer Sequence | Amplification Length/bp |
|---|---|---|
| PD-E1 | F: ATGAGAACTGGTGGTATTGGTGC | 164 |
| R: GTCACAATCTGGGCAGGTTGAAC | ||
| PyMORG1 | F: GAGCCCCAGAAGCAGACAAT | 188 |
| R: CCCGGAACATCCTCGGAAAT | ||
| PyUNC | F: GATGTCAGGGCTGCCTACG | 188 |
| R: TAAAGGCTCCTGCTCCATGTG | ||
| PyDET2 | F: TACAAGGTTCCCAAGGGTGG | 154 |
| R: AACTCCTCCCCGTCAGGTAA | ||
| PySOD1 | F: CCTCATGGATTCCACCTGCAT | 174 |
| R: TATTATTGCCTCTGCCACCCC | ||
| PyMYBP | F: GGTCCTGGGAATTGGAGAGC | 166 |
| R: TGTTGCCTAGAAGGGCTTGG | ||
| PyfrmA | F: GCAAGGGGCGGCAAAAATAA | 155 |
| R: CCACACCCAATCCACCTGTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Guo, T.; Li, H.; Ma, S.; Zhang, X.; He, C.; Zong, D. DNA Methylation Analysis of Growth Differences between Upright and Inverted Cuttings of Populus yunnanensis. Int. J. Mol. Sci. 2024, 25, 5096. https://doi.org/10.3390/ijms25105096
Guo H, Guo T, Li H, Ma S, Zhang X, He C, Zong D. DNA Methylation Analysis of Growth Differences between Upright and Inverted Cuttings of Populus yunnanensis. International Journal of Molecular Sciences. 2024; 25(10):5096. https://doi.org/10.3390/ijms25105096
Chicago/Turabian StyleGuo, Haiyang, Tiansu Guo, Hailin Li, Shaojie Ma, Xiaolin Zhang, Chengzhong He, and Dan Zong. 2024. "DNA Methylation Analysis of Growth Differences between Upright and Inverted Cuttings of Populus yunnanensis" International Journal of Molecular Sciences 25, no. 10: 5096. https://doi.org/10.3390/ijms25105096