
p20BAP31 Induces Autophagy in Colorectal Cancer Cells by Promoting PERK-Mediated ER Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overexpression of p20BAP31 Induces Autophagy
2.2. The Effect of p20BAP31 on the PI3K-AKT-mTOR Pathway
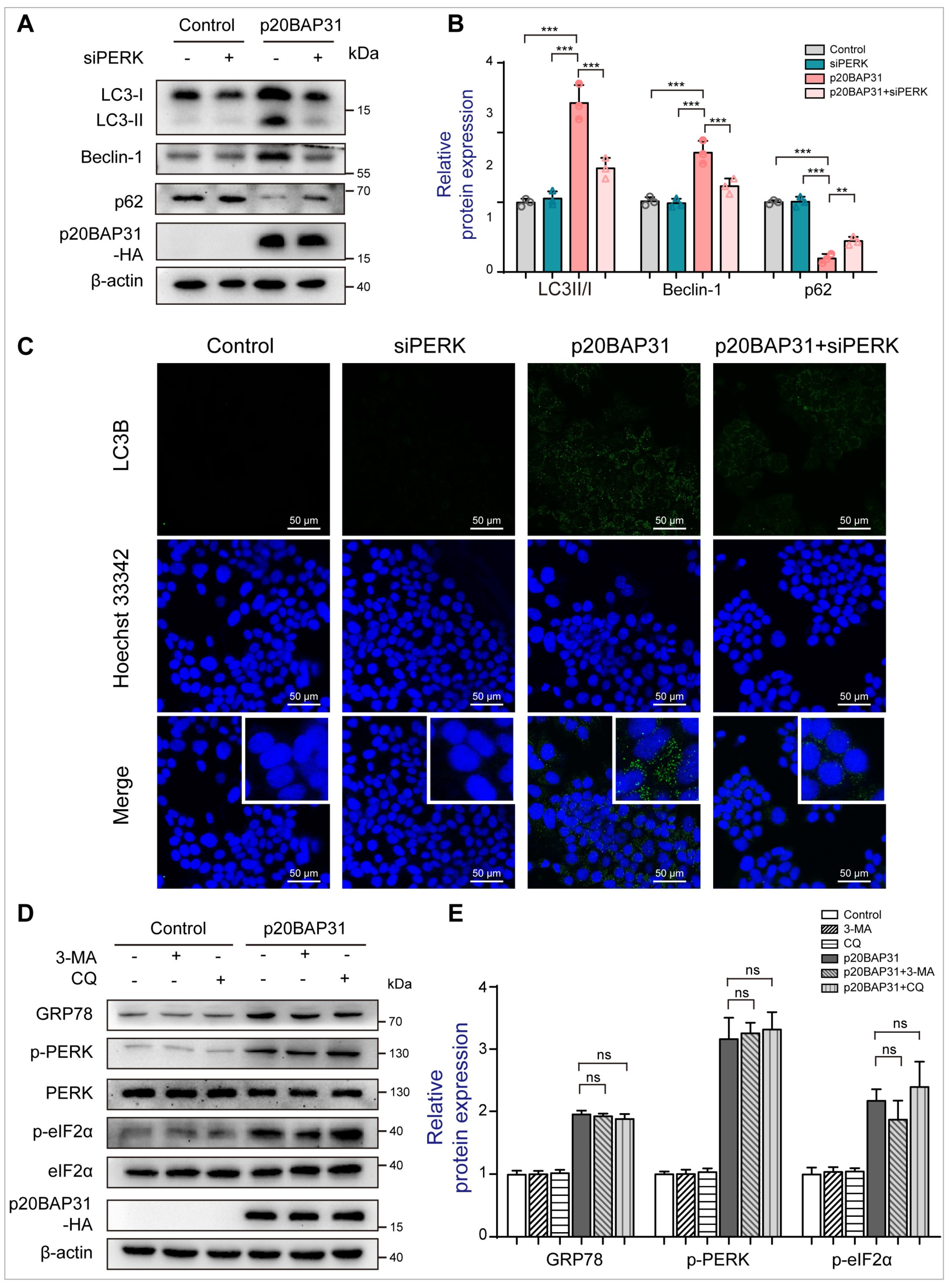
2.3. The Relationship between p20BAP31-Induced Autophagy and ER Stress
2.4. The Effect of ROS Accumulation on p20BAP31-Induced Autophagy
2.5. Analysis of Autophagy Inhibition Induced by p20BAP31 on Cell Apoptosis
2.6. Overexpression of p20BAP31 in CRC Induces Autophagy and Inhibits Tumor Growth In Vivo
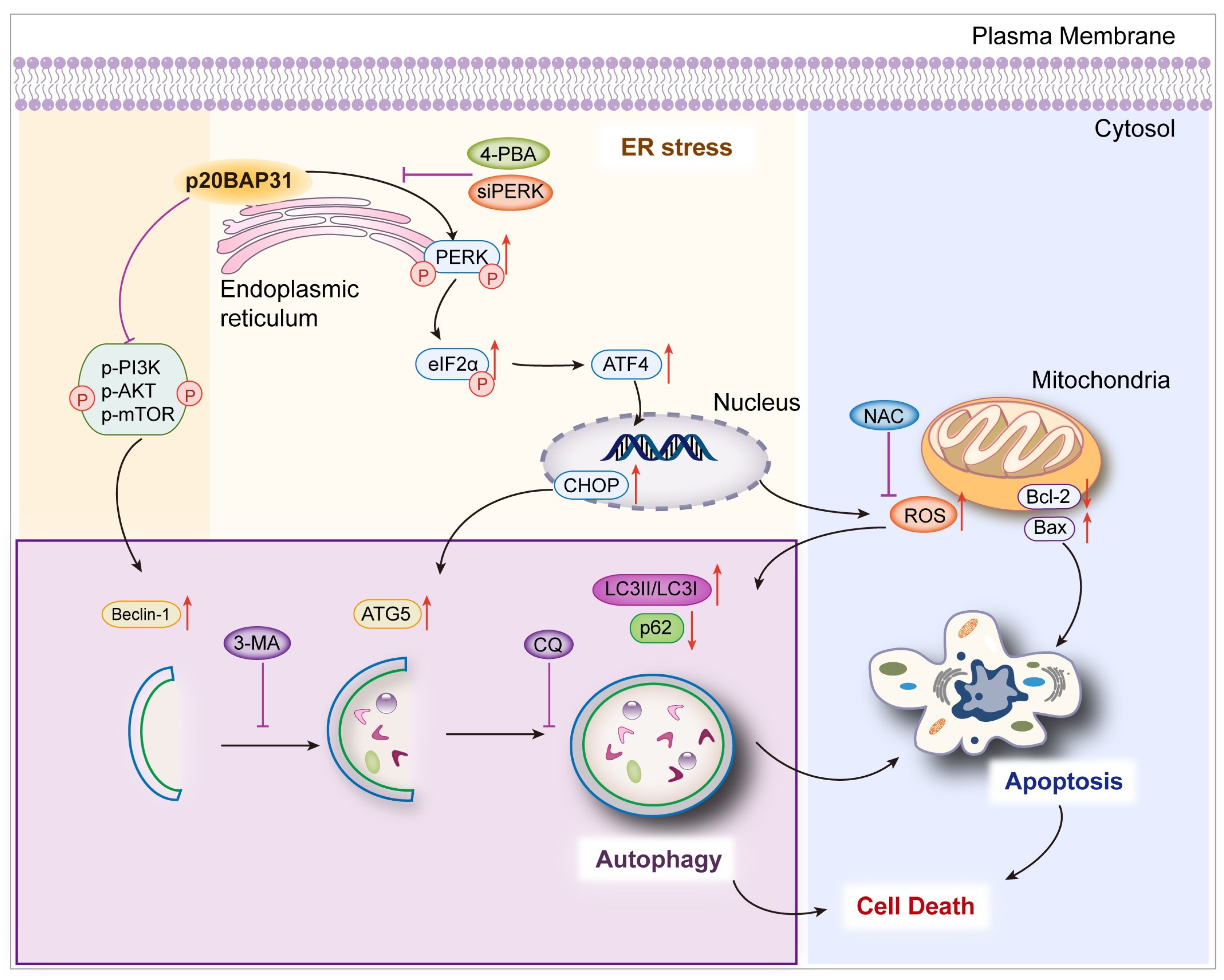
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Plasmid Construction and Transfection
4.4. Small Interfering RNA (siRNA) Transfection
4.5. Western Blot Analysis
4.6. Real-Time PCR Analysis
4.7. Immunofluorescence Assay
4.8. Cell Apoptosis Assay
4.9. Colony Forming Assay
4.10. Measurement of Intracellular ROS Levels
4.11. Tumor Xenografts
4.12. Immunohistochemistry Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adachi, T.; Schamel, W.W.; Kim, K.M.; Watanabe, T.; Becker, B.; Nielsen, P.J.; Reth, M. The specificity of association of the IgD molecule with the accessory proteins BAP31/BAP29 lies in the IgD transmembrane sequence. EMBO J. 1996, 15, 1534–1541. [Google Scholar] [CrossRef]
- Kim, K.M.; Adachi, T.; Nielsen, P.J.; Terashima, M.; Lamers, M.C.; Köhler, G.; Reth, M. Two new proteins preferentially associated with membrane immunoglobulin D. EMBO J. 1994, 13, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Zen, K.; Utech, M.; Liu, Y.; Soto, I.; Nusrat, A.; Parkos, C.A. Association of BAP31 with CD11b/CD18: Potential role in intracellular trafficking of CD11B/CD18 in neutrophils. J. Biol. Chem. 2004, 279, 44924–44930. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Skorupa, E.; Kemper, B. BAP31 Is Involved in the Retention of Cytochrome P450 2C2 in the Endoplasmic Reticulum. J. Biol. Chem. 2006, 281, 4142–4148. [Google Scholar] [CrossRef]
- Annaert, W.G.; Becker, B.; Kistner, U.; Reth, M.; Jahn, R. Export of Cellubrevin from the Endoplasmic Reticulum Is Controlled by BAP31. J. Cell Biol. 1997, 139, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; Van Prooyen, N.; Ladasky, J.J.; Edidin, M. Interaction of Bap31 and MHC Class I Molecules and Their Traffic Out of the Endoplasmic Reticulum 1. J. Immunol. 2009, 182, 4776–4783. [Google Scholar] [CrossRef]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.-Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 Complex Transduces Apoptotic Signals from Endoplasmic Reticulum to Mitochondria under Endoplasmic Reticulum Stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef]
- Ng, F.W.H.; Nguyen, M.; Kwan, T.; Branton, P.E.; Nicholson, D.W.; Cromlish, J.A.; Shore, G.C. p28 Bap31, a Bcl-2/Bcl-XL- and Procaspase-8–associated Protein in the Endoplasmic Reticulum. J. Cell Biol. 1997, 139, 327–338. [Google Scholar] [CrossRef]
- Wang, B.; Nguyen, M.; Chang, N.C.; Shore, G.C. Fis1, Bap31 and the kiss of death between mitochondria and endoplasmic reticulum. EMBO J. 2011, 30, 451–452. [Google Scholar] [CrossRef]
- Machihara, K.; Namba, T. BAP31 Inhibits Cell Adaptation to ER Stress Conditions, Negatively Regulating Autophagy Induction by Interaction with STX17. Cells 2019, 8, 1350. [Google Scholar] [CrossRef]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Heath-Engel, H.M.; Wang, B.; Shore, G.C. Bcl2 at the endoplasmic reticulum protects against a Bax/Bak-independent paraptosis-like cell death pathway initiated via p20Bap31. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 335–347. [Google Scholar] [CrossRef]
- Jiang, X.; Li, G.; Zhu, B.; Zang, J.; Lan, T.; Jiang, R.; Wang, B. p20BAP31 induces cell apoptosis via both AIF caspase-independent and the ROS/JNK mitochondrial pathway in colorectal cancer. Cell. Mol. Biol. Lett. 2023, 28, 25. [Google Scholar] [CrossRef]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 1516–1523. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Liao, Y.; Duan, B.; Zhang, Y.; Zhang, X.; Xia, B. Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. J. Biol. Chem. 2019, 294, 20009–20023. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Wang, C.-M.; Hsiao, I.H.; Liu, Y.-L.; Lin, W.-H.; Lin, C.-L.; Hung, H.-C.; Liu, G.-Y. Peptidylarginine deiminase 2 promotes T helper 17-like T cell activation and activated T cell-autonomous death (ACAD) through an endoplasmic reticulum stress and autophagy coupling mechanism. Cell. Mol. Biol. Lett. 2022, 27, 19. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Li, J.; Jiang, M.; Yu, Z.; Xiong, C.; Pan, J.; Cai, Z.; Xu, N.; Zhou, X.; Huang, Y.; Yang, Z. Artemisinin relieves osteoarthritis by activating mitochondrial autophagy through reducing TNFSF11 expression and inhibiting PI3K/AKT/mTOR signaling in cartilage. Cell. Mol. Biol. Lett. 2022, 27, 62. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Shin, S.Y.; Lee, K.S.; Choi, Y.-K.; Lim, H.J.; Lee, H.G.; Lim, Y.; Lee, Y.H. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; Medema, J.P. Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat. Rev. 2008, 34, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy 2013, 9, 626–628. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef]
- Lee, H.; Noh, J.Y.; Oh, Y.; Kim, Y.; Chang, J.W.; Chung, C.W.; Lee, S.T.; Kim, M.; Ryu, H.; Jung, Y.K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2012, 21, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, B.; Luan, P.; Gu, B.; Wan, Q.; Huang, X.; Liao, W.; Liu, J. PI3K/AKT/mTOR/p70S6K Pathway Is Involved in Aβ25-35-Induced Autophagy. Biomed Res. Int. 2015, 2015, 161020. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Ghavami, S.; Eshragi, M.; Ande, S.R.; Chazin, W.J.; Klonisch, T.; Halayko, A.J.; McNeill, K.D.; Hashemi, M.; Kerkhoff, C.; Los, M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010, 20, 314–331. [Google Scholar] [CrossRef]
- Kaminskyy, V.O.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Li, G.; Zhu, B.; Yang, J.; Cui, S.; Jiang, R.; Wang, B. p20BAP31 Induces Autophagy in Colorectal Cancer Cells by Promoting PERK-Mediated ER Stress. Int. J. Mol. Sci. 2024, 25, 5101. https://doi.org/10.3390/ijms25105101
Jiang X, Li G, Zhu B, Yang J, Cui S, Jiang R, Wang B. p20BAP31 Induces Autophagy in Colorectal Cancer Cells by Promoting PERK-Mediated ER Stress. International Journal of Molecular Sciences. 2024; 25(10):5101. https://doi.org/10.3390/ijms25105101
Chicago/Turabian StyleJiang, Xiaohan, Guoxun Li, Benzhi Zhu, Jiaying Yang, Shuyu Cui, Rui Jiang, and Bing Wang. 2024. "p20BAP31 Induces Autophagy in Colorectal Cancer Cells by Promoting PERK-Mediated ER Stress" International Journal of Molecular Sciences 25, no. 10: 5101. https://doi.org/10.3390/ijms25105101