
Lactate Dehydrogenase-Elevating Virus Infection Inhibits MOG Peptide Presentation by CD11b+CD11c+ Dendritic Cells in a Mouse Model of Multiple Sclerosis
 and
and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
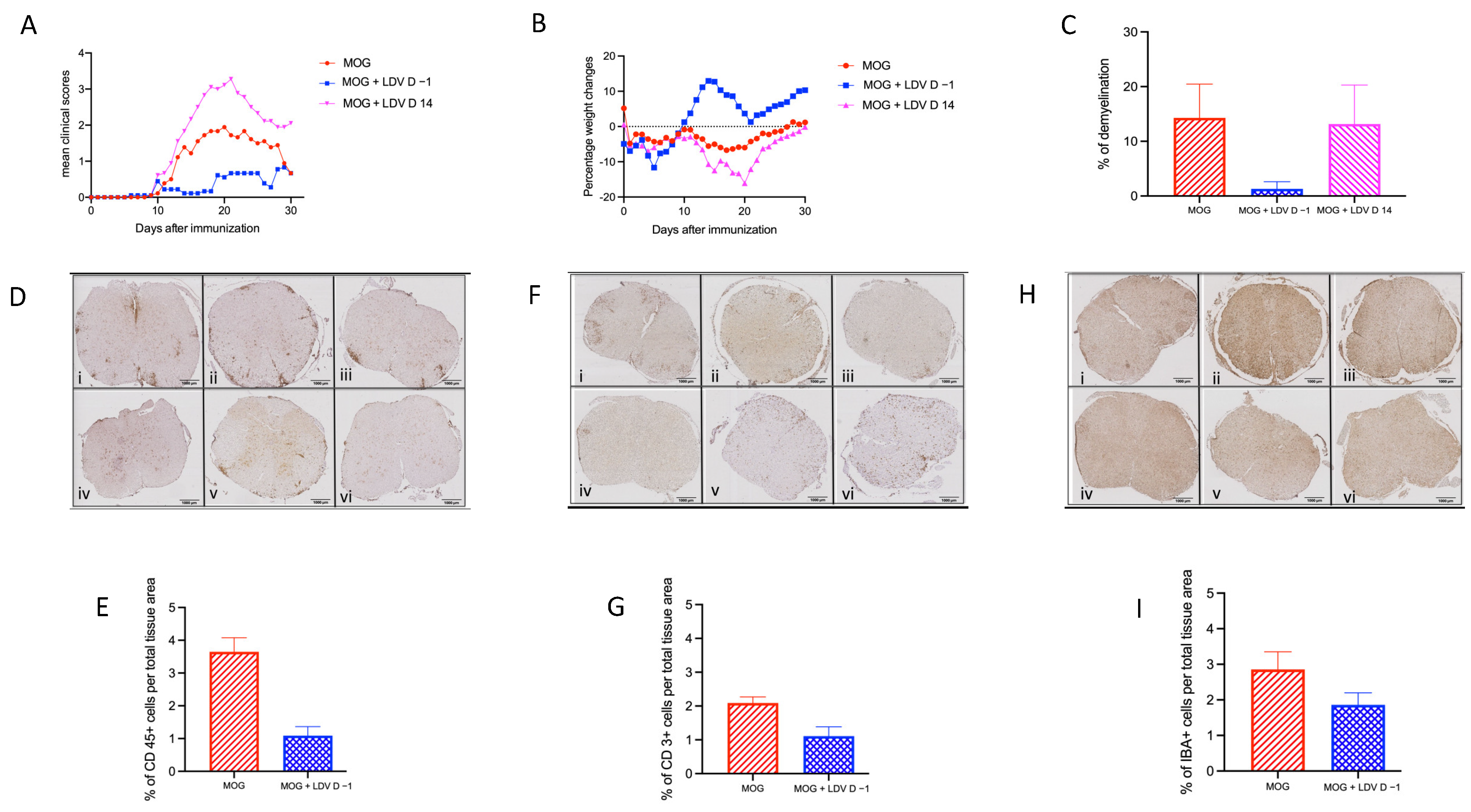
2.1. EAE Prevention by Acute LDV Infection
2.2. Inhibition of MOG Peptide Presentation by LDV Infection
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Induction and Evaluation of EAE
4.3. Viral Infection
4.4. Demyelination Analysis
4.5. Immunohistochemistry
4.6. MOG-Specific T Cell and DC Preparation
4.7. In Vitro Culture
4.8. Proliferation Assay
4.9. Cytokine Measurements
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Landry, R.L.; Embers, M.E. The probable infectious origin of multiple sclerosis. NeuroSci 2023, 4, 211–234. [Google Scholar] [CrossRef]
- Rook, G.A.W. Hygiene hypothesis and autoimmune diseases. Clin. Rev. Allerg. Immunol. 2012, 42, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-W.; Jun, H.-S. Viruses cause type 1 diabetes in animals. Ann. N.Y. Acad. Sci. 2006, 1079, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lehmann, P.V.; Kaufman, D.L. T cell cross-reactivity between coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J. Exp. Med. 1994, 180, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, M.S.; Ilic, A.; Fine, C.; Balasa, B.; Sarvetnick, N. Coxsackieviral-mediated diabetes: Induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin. Immunol. 2004, 110, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar]
- Hosking, M.P.; Lane, T.E. The pathogenesis of murine coronavirus infection of the central nervous system. Crit. Rev. Immunol. 2010, 30, 119–130. [Google Scholar] [CrossRef]
- Meite, M.; Leonard, S.; El Azami El Idrissi, M.; Izui, S.; Masson, P.L.; Coutelier, J.-P. Exacerbation of autoantibody-mediated hemolytic anemia by viral infection. J. Virol. 2000, 74, 6045–6049. [Google Scholar] [CrossRef]
- Musaji, A.; Cormont, F.; Thirion, G.; Cambiaso, C.L.; Coutelier, J.-P. Exacerbation of autoantibody-mediated thrombocytopenic purpura by infection with mouse viruses. Blood 2004, 104, 2102–2106. [Google Scholar] [CrossRef]
- Oldstone, M.B.A.; Dixon, F.J. Inhibition of antibodies to nuclear antigen and to DNA in New Zealand mice infected with lactate dehydrogenase virus. Science 1972, 175, 784–786. [Google Scholar] [CrossRef]
- Takei, I.; Asaba, Y.; Kasatani, T.; Maruyama, T.; Watanabe, K.; Yanagawa, T.; Saruta, T.; Ishii, T. Suppression of development of diabetes in NOD mice by lactate dehydrogenase virus infection. J. Autoimmun. 1992, 5, 665–673. [Google Scholar] [CrossRef]
- Inada, T.; Mims, C.A. Infection of mice with lactic dehydrogenase virus prevents development of experimental allergic encephalomyelitis. J. Neuroimmunol. 1986, 11, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.G.; Bernard, C.C. The structure and function of myelin oligodendrocyte glycoprotein. J. Neurochem. 1999, 72, 1–9. [Google Scholar] [CrossRef]
- Frausto, R.F.; Crocker, S.J.; Eam, B.; Whitmire, J.K.; Whitton, J.L. Myelin oligodendrocyte glycoprotein peptide-induced experimental allergic encephalomyelitis and T cell responses are unaffected by immunoproteasome deficiency. J. Neuroimmunol. 2007, 192, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Gaignage, M.; Marillier, R.G.; Uyttenhove, C.; Dauguet, N.; Saxena, A.; Ryffel, B.; Michiels, T.; Coutelier, J.-P.; Van Snick, J. Mouse nidovirus LDV infection alleviates graft versus host disease and induces type I FN-dependent inhibition of dendritic cells and allo-responsive T cells. Immun. Inflamm. Dis. 2017, 5, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Gaignage, M.; Marillier, R.G.; Cochez, P.M.; Dumoutier, L.; Uyttenhove, C.; Coutelier, J.-P.; Van Snick, J. The TLR7 ligand R848 prevents mouse graft-versus-host disease and cooperates with anti-interleukin-27 antibody for maximal protection and regulatory T-cell upregulation. Haematologica 2019, 104, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Wlodarczyk, M.F.; Benamar, M.; Bassot, E.; Salvioni, A.; Kassem, S.; Berry, A.; Saoudi, A.; Blanchard, N. A virus hosted in malaria-infected blood protects against T cell-mediated inflammatory diseases by impairing DC function in a type I IFN-dependent manner. mBio 2020, 11, e03394-19. [Google Scholar] [CrossRef]
- Romagnani, S. The role of lymphocytes in allergic disease. J. Allergy Clin. Immunol. 2000, 105, 399–408. [Google Scholar] [CrossRef]
- Ndoricyimpaye, E.L.; Van Snick, J.; Niyoyita, J.d.D.; Kanimba, P.; Mbonimpa, J.B.; Rutayisire, R.; Rutayisire, R.; Ndahindwa, V.; Cheou, P.; Coutelier, J.-P.; et al. Integrated analysis of cytokine profiles in malaria patients disclose selective upregulation of TGF-β1, β3, and IL-9 in mild clinical presentation. Int. J. Mol. Sci. 2022, 23, 12665. [Google Scholar] [CrossRef]
- Yazdanbakhsh, M.; Kremsner, P.G.; van Ree, R. Allergy, parasites, and the hygiene hypothesis. Science 2002, 296, 490–494. [Google Scholar] [CrossRef]
- Giles, D.A.; Duncker, P.C.; Wilkinson, N.M.; Washnock-Schmid, J.M.; Segal, B.J. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J. Clin. Investig. 2018, 128, 5322–5334. [Google Scholar] [CrossRef]
- Yadav, P.K.; Chandrakar, P.; Sharma, P.; Vishwakarma, P.; Parmar, N.; Srivastava, M.; Kar, S. Reciprocal changes in CD11c+CD11b+ and CD11c+CD8α+ dendritic cell subsets determine protective or permissive immune response in murine experimental VL. Vaccine 2020, 38, 355–365. [Google Scholar] [CrossRef]
- Ammann, C.G.; Messer, R.J.; Peterson, K.E.; Hasenkrug, K.J. Lactate dehydrogenase-elevating virus induces systemic lymphocyte activation via TLR7-dependent IFNalpha responses by plasmacytoid dendritic cells. PLoS ONE 2009, 4, e6105. [Google Scholar] [CrossRef]
- Isakov, N.; Feldman, M.; Segal, S. Acute infection of mice with acute dehydrogenase virus (LDV) impairs the antigen-presenting capacity of their macrophages. Cell. Immunol. 1982, 66, 317–332. [Google Scholar] [CrossRef]
- Li, H.; Zhang, G.-X.; Chen, Y.; Xu, H.; Fitzgerald, D.C.; Zhao, Z.; Rostami, A. CD11c+CD11b+ dendritic cells play an important role in intravenous tolerance and the suppression of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 181, 2483–2493. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soe, P.P.; Gaignage, M.; Mandour, M.F.; Marbaix, E.; Van Snick, J.; Coutelier, J.-P. Lactate Dehydrogenase-Elevating Virus Infection Inhibits MOG Peptide Presentation by CD11b+CD11c+ Dendritic Cells in a Mouse Model of Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 4950. https://doi.org/10.3390/ijms25094950
Soe PP, Gaignage M, Mandour MF, Marbaix E, Van Snick J, Coutelier J-P. Lactate Dehydrogenase-Elevating Virus Infection Inhibits MOG Peptide Presentation by CD11b+CD11c+ Dendritic Cells in a Mouse Model of Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(9):4950. https://doi.org/10.3390/ijms25094950
Chicago/Turabian StyleSoe, Pyone Pyone, Mélanie Gaignage, Mohamed F. Mandour, Etienne Marbaix, Jacques Van Snick, and Jean-Paul Coutelier. 2024. "Lactate Dehydrogenase-Elevating Virus Infection Inhibits MOG Peptide Presentation by CD11b+CD11c+ Dendritic Cells in a Mouse Model of Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 9: 4950. https://doi.org/10.3390/ijms25094950