
Effect of Root-Knot Nematode Disease on Bacterial Community Structure and Diversity in Peanut Fields
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Samples Collection and Processing
2.2. 16S rDNA Full-Length Amplification and Sequencing
2.3. Determination of Soil Physical and Chemical Properties
2.4. Statistical Analysis
3. Results
3.1. Bacterial Community Diversity of Bulk Soil in Peanuts Field
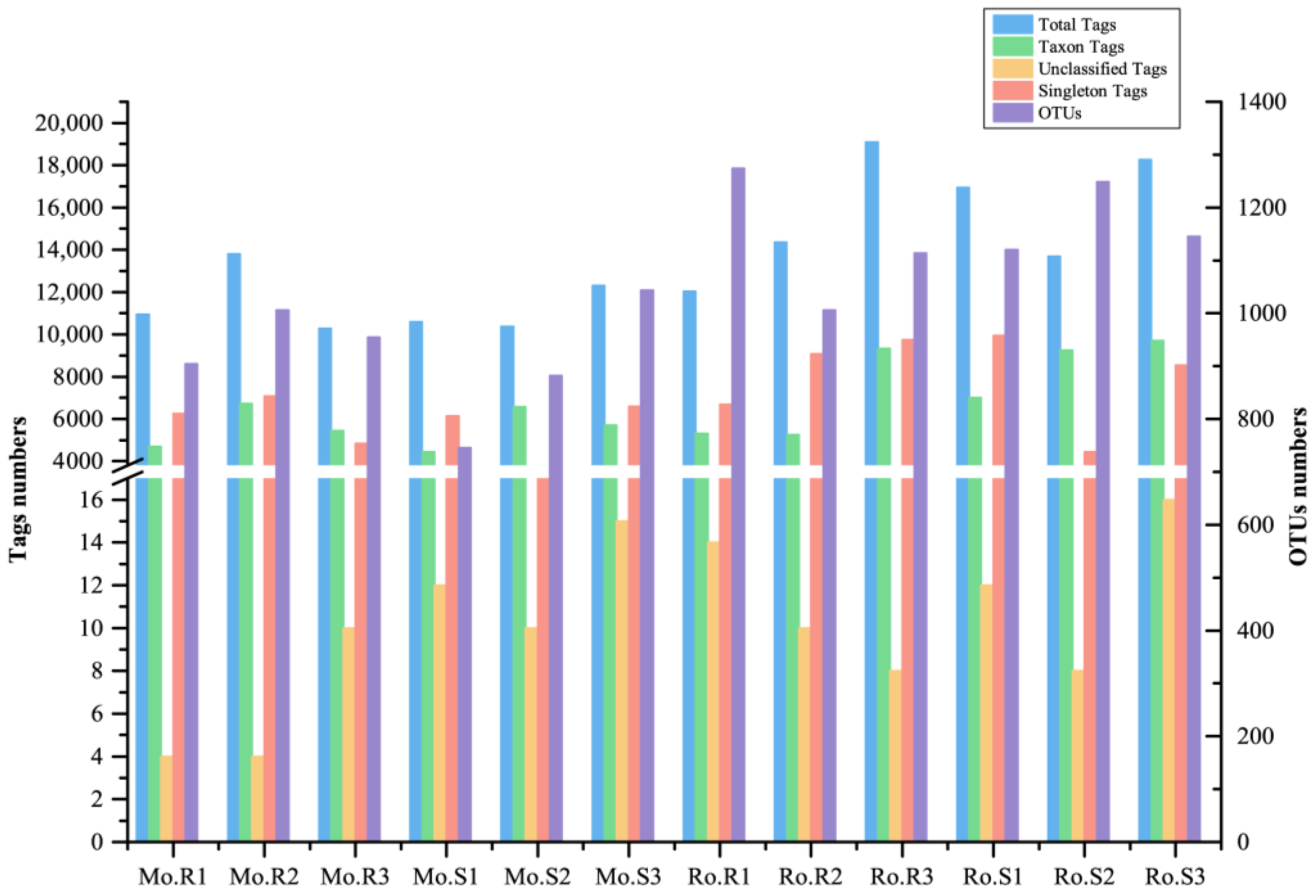
3.1.1. Sequence Data of 16S Full-Length rDNA
3.1.2. Alpha Diversity
3.1.3. Beta Diversity
3.1.4. Bacterial Community Structure of Peanut Bulk Soil Influenced by RKN
3.2. Analysis of the Microbiological Biomarkers in the Peanut Bulk Soil
3.3. Relationship between Bacterial Community Structure and Environment Factors in Peanut Bulk Soil
3.4. Analysis of Beneficial and Harmful Bacteria in the Peanut Bulk Soil
3.5. Prediction of Bacterial Functional Potential in the Peanut Bulk Soil
4. Discussion
4.1. Effect of Severe RKN Disease on the Bacterial Diversity in Peanut Bulk Soil
4.2. Differential Bacterial Communities between Peanut Bulk Soils of Resistant and Susceptible to RKN
4.3. The Role of Environment Factors in Bacterial Communities of Peanut Bulk Soil
4.4. Bacterial Potential Function in Peanut Bulk Soil
4.5. Beneficial and Harmful Bacteria in Peanut Bulk Soil
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Launio, C.C.; Luis, J.S.; Angeles, Y.B. Factors influencing adoption of selected peanut protection and production technologies in Northern Luzon, Philippines. Technol. Soc. 2018, 55, 56–62. [Google Scholar] [CrossRef]
- Settaluri, V.S.; Kandala, C.V.K.; Puppala, N.; Sundaram, J. Peanuts and their nutritional aspects-a review. Food Nutr. Sci. 2012, 3, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Toomer, O.T. Nutritional chemistry of the peanut (Arachis hypogaea). Crit. Rev. Food Sci. Nutr. 2018, 58, 3042–3053. [Google Scholar] [CrossRef]
- Toomer, O.T. A comprehensive review of the value-added uses of peanut (Arachis hypogaea) skins and by-products. Crit. Rev. Food Sci. Nutr. 2020, 60, 341–350. [Google Scholar] [CrossRef]
- Osman, H.A.; Ameen, H.H.; Mohamed, M.; Elkelany, U.S. Efficacy of integrated microorganisms in controlling root-knot nematode Meloidogyne javanica infecting peanut plants under field conditions. Bull. Natl. Res. Cent. 2020, 44, 134. [Google Scholar] [CrossRef]
- Nagy, E.D.; Chu, Y.; Guo, Y.; Khanal, S.; Tang, S.; Li, Y.; Dong, W.B.; Timper, P.; Taylor, C.; Ozias-Akins, P.; et al. Recombination is suppressed in an alien introgression in peanut harboring Rma, a dominant root-knot nematode resistance gene. Mol. Breed. 2010, 26, 357–370. [Google Scholar] [CrossRef]
- Tirumalaraju, S.V.; Jain, M.; Gallo, M. Differential gene expression in roots of nematode-resistant and-susceptible peanut (Arachis hypogaea) cultivars in response to early stages of peanut root-knot nematode (Meloidogyne arenaria) parasitization. J. Plant Physiol. 2011, 168, 481–492. [Google Scholar] [CrossRef]
- Ballén-Taborda, C.; Chu, Y.; Ozias-Akins, P.; Timper, P.; Jackson, S.A.; Bertioli, D.J.; Leal-Bertioli, S.C. Validation of resistance to root-knot nematode incorporated in peanut from the wild relative Arachis stenosperma. Agron. J. 2021, 113, 2293–2302. [Google Scholar] [CrossRef]
- Ballén-Taborda, C.; Chu, Y.; Ozias-Akins, P.; Timper, P.; Holbrook, C.C.; Jackson, S.A.; Bertioli, D.J.; Leal-Bertioli, S.C.M. A new source of root-knot nematode resistance from Arachis stenosperma incorporated into allotetraploid peanut (Arachis hypogaea). Sci. Rep. 2019, 9, 17702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Deng, X.; Gao, L.; Cai, X.; Yan, D.; Cai, Y.; Chen, X.; Yang, M.; Tong, W.; Yu, L. Effect of marigold (Tagetes erecta L.) on soil microbial communities in continuously cropped tobacco fields. Sci. Rep. 2022, 12, 19632. [Google Scholar] [CrossRef]
- Krupinsky, J.M.; Bailey, K.L.; Mcmullen, M.P.; Gossen, B.D.; Turkington, T.K. Managing plant disease risk in diversified cropping systems. Agron. J. 2002, 94, 198–209. [Google Scholar] [CrossRef]
- Ramakrishna, W.; Yadav, R.; Li, K. Plant growth promoting bacteria in agriculture: Two sides of a coin. Appl. Soil Ecol. 2019, 138, 10–18. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, Z.-X.; Yang, D.-M.; Lu, N.; Yu, S.-Z.; Meng, J.-Y.; Chen, X.-J. Tobacco root microbial community composition significantly associated with root-knot nematode infections: Dynamic changes in microbiota and growth stage. Front. Microbiol. 2022, 13, 807057. [Google Scholar] [CrossRef]
- Lu, P.; Shi, H.; Tao, J.; Jin, J.; Wang, S.; Zheng, Q.; Liu, P.; Xiang, B.; Chen, Q.; Xu, Y.; et al. Metagenomic insights into the changes in the rhizosphere microbial community caused by the root-knot nematode Meloidogyne incognita in tobacco. Environ. Res. 2023, 216, 114848. [Google Scholar] [CrossRef]
- Li, Y.; Lei, S.; Cheng, Z.; Jin, L.; Zhang, T.; Liang, L.-M.; Cheng, L.; Zhang, Q.; Xu, X.; Lan, C.; et al. Microbiota and functional analyses of nitrogen-fixing bacteria in root-knot nematode parasitism of plants. Microbiome 2023, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Rani, P.; Singh, M.; Prashad, H.; Sharma, M. Evaluation of bacterial formulations as potential biocontrol agents against the southern root-knot nematode, Meloidogyne incognita. Egypt. J. Biol. Pest Control 2022, 32, 29. [Google Scholar] [CrossRef]
- Ullah, A.; Akbar, A.; Luo, Q.; Khan, A.H.; Manghwar, H.; Shaban, M.; Yang, X. Microbiome diversity in cotton rhizosphere under normal and drought conditions. Microb. Ecol. 2019, 77, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zhang, G.; Yu, Z.; Ding, H.; Xu, Y.; Zhang, Z. Effect of drought stress and developmental stages on microbial community structure and diversity in peanut rhizosphere soil. Int. J. Mol. Sci. 2019, 20, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Zhao, D.; Cai, C.; Song, D.; Shen, J.; Xu, A.; Qiao, Y.; Ran, Z.; Zheng, Q. Low-dose penicillin exposure in early life decreases Th17 and the susceptibility to DSS colitis in mice through gut microbiota modification. Sci. Rep. 2017, 7, 43662. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y. Bioinformatic and Statistical Analysis of Microbiome Data. In Statistical Genomics, 10th ed.; Fridley, B., Wang, X., Eds.; Springer US: New York, NY, USA, 2023; Volume 2629, pp. 183–229. [Google Scholar]
- Maughan, H.; Wang, P.W.; Diaz Caballero, J.; Fung, P.; Gong, Y.; Donaldson, S.L.; Yuan, L.; Keshavjee, S.; Zhang, Y.; Yau, Y.C.; et al. Analysis of the cystic fibrosis lung microbiota via serial Illumina sequencing of bacterial 16S rRNA hypervariable regions. PLoS ONE 2012, 7, e45791. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef] [Green Version]
- Gamliel, A.; Austerweil, M.; Kritzman, G. Non-chemical approach to soilborne pest management–organic amendments. Crop Prot. 2000, 19, 847–853. [Google Scholar] [CrossRef]
- Bulluck, L.R.; Brosius, M.; Evanylo, G.K.; Ristaino, J.B. Organic and synthetic fertility amendments influence soil microbial, physical and chemical properties on organic and conventional farms. Appl. Soil Ecol. 2002, 19, 147–160. [Google Scholar] [CrossRef]
- Bakker, M.G.; Glover, J.D.; Mai, J.G.; Kinkel, L.L. Plant community effects on the diversity and pathogen suppressive activity of soil streptomycetes. Appl. Soil Ecol. 2010, 46, 35–42. [Google Scholar] [CrossRef]
- Bao, S.D. Agricultural and Chemistry Analysis of Soil, 3rd ed.; Agriculture Press: Beijing, China, 2005; pp. 11–18. [Google Scholar]
- Olsen, S.R.; Watanabe, F.S. A method to determine a phosphorus adsorption maximum of soils as measured by the langmuir isotherm. Soil Sci. Soc. Am. J. 1957, 21, 144–149. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Cordero, I.; Balaguer, L.; Rincón, A.; Pueyo, J.J. Inoculation of tomato plants with selected PGPR represents a feasible alternative to chemical fertilization under salt stress. J. Plant Nutr. Soil Sci. 2018, 181, 694–703. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; Zhang, H.; Wei, G.; Li, Z. Beneficial bacteria activate nutrients and promote wheat growth under conditions of reduced fertilizer application. BMC Microbiol. 2020, 20, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Chen, X.; Li, Z.; Wang, M.; Che, Y.; Zhang, L.; Jiang, Z.; Jie, S. Effects of continuous cropping on bacterial community and diversity in rhizosphere soil of industrial hemp: A five-year experiment. Diversity 2022, 14, 250. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, J.; Li, T.; Liao, Y. Response of soil fungal communities to continuous cropping of flue-cured tobacco. Sci. Rep. 2020, 10, 19911. [Google Scholar] [CrossRef]
- Lakshmanan, V.; Selvaraj, G.; Bais, H.P. Functional soil microbiome: Belowground solutions to an aboveground problem. Plant Physiol. 2014, 166, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; Da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loque, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wang, X.; Zhang, W.; Zhou, Z.; Ding, C.; Liao, Y.; Li, X. Persistent organic fertilization reinforces soil-borne disease suppressiveness of rhizosphere bacterial community. Plant Soil. 2020, 452, 313–328. [Google Scholar] [CrossRef]
- Shen, Z.; Penton, C.R.; Lv, N.; Xue, C.; Yuan, X.; Ruan, Y.; Li, R.; Shen, Q. Banana Fusarium Wilt Disease Incidence Is Influenced by Shifts of Soil Microbial Communities Under Different Monoculture Spans. Microb. Ecol. 2018, 75, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Wu, X.-Q.; Xu, Y.; Wang, B.-L.; Liu, S.; Niu, J.-F.; Wang, Z. Microbial diversity and community structure changes in the rhizosphere soils of Atractylodes lancea from different planting years. Plant Signal. Behav. 2021, 16, 1854507. [Google Scholar] [CrossRef] [PubMed]
- Sasse, J.; Martinoia, E.; Northen, T. Feed your friends: Do plant exudates shape the root microbiome? Trends Plant Sci. 2017, 23, 25–41. [Google Scholar] [CrossRef] [Green Version]
- Selmar, D.; Kleinwachter, M. Stress enhances the synthesis of secondary plant products: The impact of stress-related over-reduction on the accumulation of natural products. Plant Cell Physiol. 2013, 54, 817–826. [Google Scholar] [CrossRef]
- Njira, K. Soil management practices that improve soil health: Elucidating their implications on biological indicators. J. Anim. Plant Sci. 2013, 18, 2750–2760. [Google Scholar]
- Dorr de Quadros, P.; Zhalnina, K.; Davis-Richardson, A.; Fagen, J.R.; Drew, J.; Bayer, C.; Camargo, F.A.O.; Triplett, E.W. The Effect of Tillage System and Crop Rotation on Soil Microbial Diversity and Composition in a Subtropical Acrisol. Diversity 2012, 4, 375–395. [Google Scholar] [CrossRef]
- Cao, Y. Characteristics of Rhizobacterial Community to Root-Knot Nematode Infected Tobacco and Diversity of Bacterial Flora Associated with Meloidogyne incongnita. Ph.D. Thesis, Yunan University, Kunming, China, 2015. [Google Scholar]
- Kojima, H.; Kanda, M.; Umezawa, K.; Fukui, M. Sulfurimicrobium lacus gen. nov., sp. nov., a sulfur oxidizer isolated from lake water, and review of the family Sulfuricellaceae to show that it is not a later synonym of Gallionellaceae. Arch. Microbiol. 2021, 203, 317–323. [Google Scholar] [CrossRef]
- Watanabe, T.; Kojima, H.; Fukui, M. Sulfuriferula multivorans gen. nov., sp. nov., isolated from a freshwater lake, reclassification of ‘Thiobacillus plumbophilus’ as Sulfuriferula plumbophilus sp. nov., and description of Sulfuricellaceae fam. nov. and Sulfuricellales ord. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 1504–1508. [Google Scholar] [CrossRef] [Green Version]
- Kulichevskaya, I.S.; Detkova, E.N.; Bodelier, P.L.E.; Rijpstra, W.I.C.; Sinninghe Damste, J.S.; Dedysh, S.N. Singulisphaera rosea sp. nov., a planctomycete from acidic Sphagnum peat, and emended description of the genus Singulisphaera. Int. J. Syst. Evol. Microbiol. 2012, 62, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Costa, O.Y.A.; De Hollander, M.; Pijl, A.; Liu, B.; Kuramae, E.E. Cultivation-independent and cultivation-dependent metagenomes reveal genetic and enzymatic potential of microbial community involved in the degradation of a complex microbial polymer. Microbiome 2020, 8, 76. [Google Scholar] [CrossRef]
- De Vries, R.P.; Kester, H.C.; Poulsen, C.H.; Benen, J.A.; Visser, J. Synergy between enzymes from Aspergillus involved in the degradation of plant cell wall polysaccharides. Carbohydr. Res. 2000, 327, 401–410. [Google Scholar] [CrossRef]
- Huang, G.; Dong, R.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. Developmental expression and molecular analysis of two Meloidogyne incognita pectate lyase genes. Int. J. Parasitol. 2005, 35, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Kolb, E.; Legue, V.; Bogeat-Triboulot, M.B. Physical root-soil interactions. Phys. Biol. 2017, 14, 065004. [Google Scholar] [CrossRef] [PubMed]
- Andrew, D.R.; Fitak, R.R.; Munguia-Vega, A.; Racolta, A.; Martinson, V.G.; Dontsova, K. Abiotic factors shape microbial diversity in Sonoran Desert soils. Appl. Environ. Microbiol. 2012, 78, 7527–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chen, Y.M.; He, R.L.; Deng, C.C.; Liu, J.W.; Liu, Y. Responses of soil microbial community structure and function to simulated warming in alpine forest. J. Appl. Ecol. 2016, 27, 2855–2863. [Google Scholar]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Subbarao, G.V.; Yoshihashi, T.; Worthington, M.; Nakahara, K.; Ando, Y.; Sahrawat, K.L.; Rao, I.M.; Lata, J.-C.; Kishii, M.; Braun, H.-J. Suppression of soil nitrification by plants. Plant Sci. 2015, 233, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Niu, Q.; Han, Y.; Xu, L. Effects of crop rotation on soil physicochemical properties and bacterial community of foxtail millet rhizosphere soil. J. Agro-Environ. Sci. 2018, 37, 2802–2809. [Google Scholar]
- Jiao, X.-L.; Zhang, X.-S.; Lu, X.-H.; Qin, R.; Bi, Y.-M.; Gao, W.-W. Effects of maize rotation on the physicochemical properties and microbial communities of American ginseng cultivated soil. Sci. Rep. 2019, 9, 8615. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.M.; Lee, J.S.; Bae, H.M.; Yi, T.H.; Kim, S.Y.; Lee, S.T.; Im, W.T. Inquilinus ginsengisoli sp. nov., isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 2011, 61, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.H.; Lee, J.S.; An, D.S.; Whon, T.W.; Kim, S.G. Nocardioides panacisoli sp. nov., isolated from the soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 2010, 60, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, K.; Xing, R.; Liu, S.; Chen, X.; Yang, H.; Li, P. miRNA and mRNA expression profiles reveal insight into the chitosan-mediated regulation of plant growth. J. Agric Food Chem. 2018, 66, 3810–3822. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yu, M.; Hou, R.; Li, L.; Ren, X.; Jiao, C.; Yang, L.; Xu, H. Changes in the soil microbial community are associated with the occurrence of Panax quinquefolius L. root rot diseases. Plant Soil. 2019, 438, 143–156. [Google Scholar] [CrossRef]
- Vessey, J.K. Plant growth promoting rhizobacteria as biofertilizers. Plant Soil. 2003, 255, 571–586. [Google Scholar] [CrossRef]
- Wang, B.; Li, R.; Ruan, Y.; Ou, Y.; Zhao, Y.; Shen, Q. Pineapple–banana rotation reduced the amount of Fusarium oxysporum more than maize–banana rotation mainly through modulating fungal communities. Soil Biol. Biochem. 2015, 86, 77–86. [Google Scholar] [CrossRef]
- Zuo, W.; Song, B.; Shi, Y.; Zupanic, A.; Guo, S.; Huang, H.; Jiang, L.; Yu, Y. Using Bacillus thuringiensis HM-311@hydroxyapatite@biochar beads to remediate Pb and Cd contaminated farmland soil. Chemosphere 2022, 307, 135797. [Google Scholar] [CrossRef]
- Basumatary, B.; Das, D.; Choudhury, B.; Dutta, P.; Bhattacharyya, A. Isolation and characterization of endophytic bacteria from tomato foliage and their in vitro efficacy against root-knot nematodes. J. Nematol. 2021, 53, 1–16. [Google Scholar] [CrossRef]
- Azarias Guimaraes, A.; Duque Jaramillo, P.M.; Simao Abrahao Nobrega, R.; Florentino, L.A.; Barroso Silva, K.; De Souza Moreira, F.M. Genetic and symbiotic diversity of nitrogen-fixing bacteria isolated from agricultural soils in the western Amazon by using cowpea as the trap plant. Appl. Environ. Microbiol. 2012, 78, 6726–6733. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.R.; Schisler, D.A.; Ryder, M.H.; Adkins, P.G. Bacteria suppress damping-off caused by Pythium ultimumvar. Sporangiiferum, and promote growth, in bedding plants. Soil Biol. Biochem. 1994, 26, 1431–1437. [Google Scholar] [CrossRef]
- Lee, S.-W.; Cooksey Donald, A. Genes Expressed in Pseudomonas putidaduring Colonization of a Plant-Pathogenic Fungus. Appl. Environ. Microbiol. 2000, 66, 2764–2772. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Mo.S | Mo.R | Ro.S | Ro.R |
|---|---|---|---|---|
| pH | 4.5 ± 0.1 a | 4.6 ± 0.1 a | 4.8 ± 0.2 a | 5.1 ± 0.2 a |
| Organic matter (OM, g/kg) | 13.5 ± 0.7 a | 14.9 ± 0.7 a | 12.1 ± 0.3 a | 10.9 ± 0.7 a |
| Available nitrogen (AN, mg/kg) | 82.9 ± 10.8 a | 85.5 ± 10.4 a | 60.7 ± 5.7 a | 54.6 ± 6.3 a |
| Rapidly available phosphorus (AP, mg/kg) | 103.4 ± 8.2 a | 121.1 ± 13.3 a | 94.3 ± 1.3 a | 81.1 ± 6.3 a |
| Available potassium (AK, mg/kg) | 46.1 ± 0.0 a | 53.6 ± 14.2 a | 61.2 ± 3.3 a | 42.3 ± 6.5 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Ren, Y.; Zhang, X.; Chen, G.; Wang, C.; Wu, Q.; Li, S.; Zhan, F.; Sheng, L.; Wei, W.; et al. Effect of Root-Knot Nematode Disease on Bacterial Community Structure and Diversity in Peanut Fields. Agronomy 2023, 13, 1803. https://doi.org/10.3390/agronomy13071803
Wu L, Ren Y, Zhang X, Chen G, Wang C, Wu Q, Li S, Zhan F, Sheng L, Wei W, et al. Effect of Root-Knot Nematode Disease on Bacterial Community Structure and Diversity in Peanut Fields. Agronomy. 2023; 13(7):1803. https://doi.org/10.3390/agronomy13071803
Chicago/Turabian StyleWu, Lijun, Yan Ren, Xiangsong Zhang, Guanghui Chen, Chuantang Wang, Qi Wu, Shuangling Li, Fudong Zhan, Li Sheng, Wenliang Wei, and et al. 2023. "Effect of Root-Knot Nematode Disease on Bacterial Community Structure and Diversity in Peanut Fields" Agronomy 13, no. 7: 1803. https://doi.org/10.3390/agronomy13071803