

A Proteomic Approach Identified TFEB as a Key Player in the Protective Action of Novel CB2R Bitopic Ligand FD22a against the Deleterious Effects Induced by β-Amyloid in Glial Cells

 , , , , and
, , , , and 
Abstract
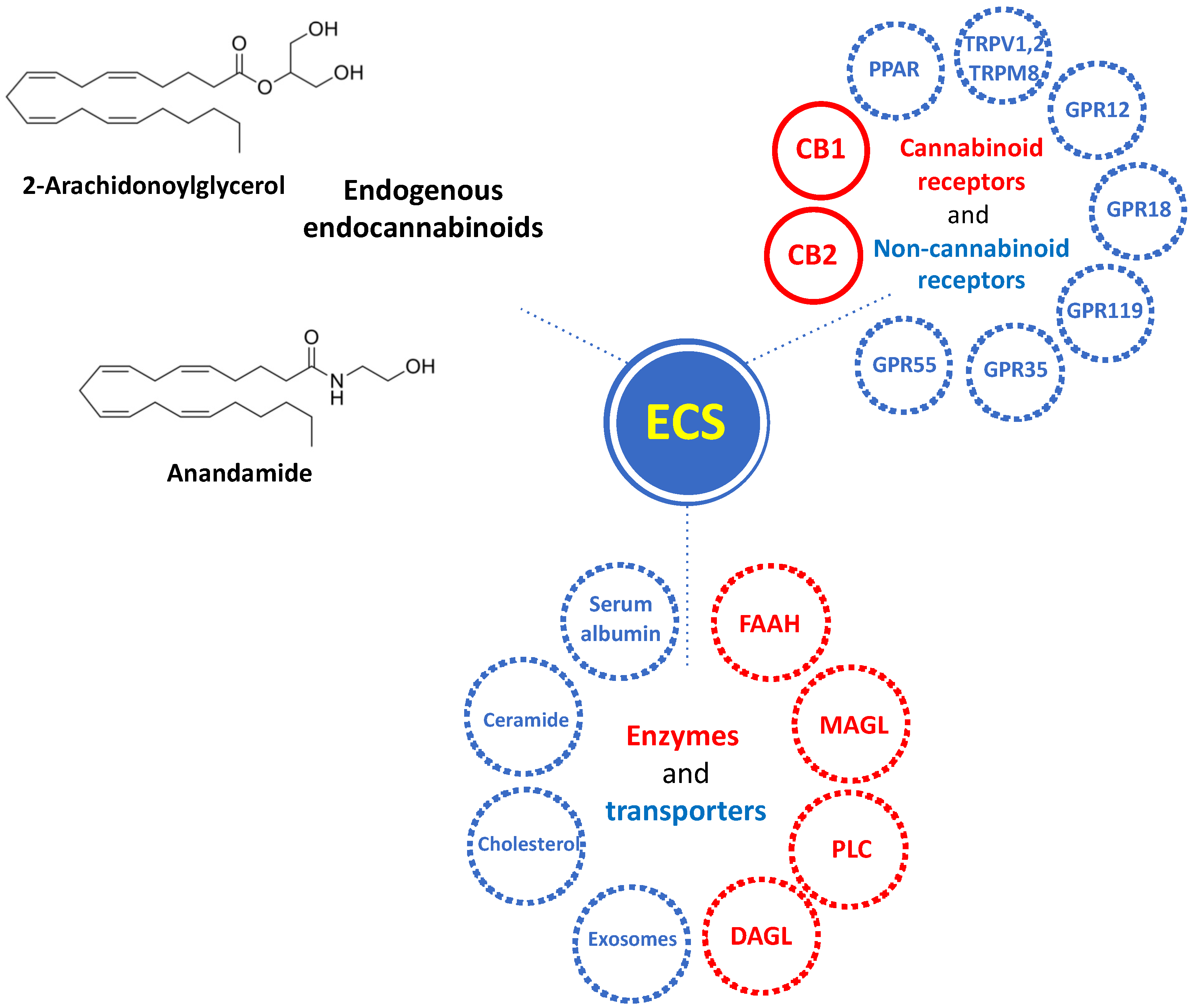
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures, Reagents, and Treatments
2.1.1. MTT (Cell Viability Assay)
2.1.2. Release of Inflammatory Cytokines
2.1.3. Gene Expression Analysis
2.2. Proteomic Analysis
2.2.1. In Gel Digestion and Mass Spectrometry
2.2.2. Bioinformatic Analysis
2.2.3. Western Blot Analysis
2.2.4. Statistical Analysis
3. Results
3.1. FD22a Prevents Aβ25–35-Induced Cytotoxicity in HMC3 and U87-MG Cells
3.2. FD22a Inhibits β-Amyloid-Mediated Release of Proinflammatory Factors in HMC3 and U87-MG Cells
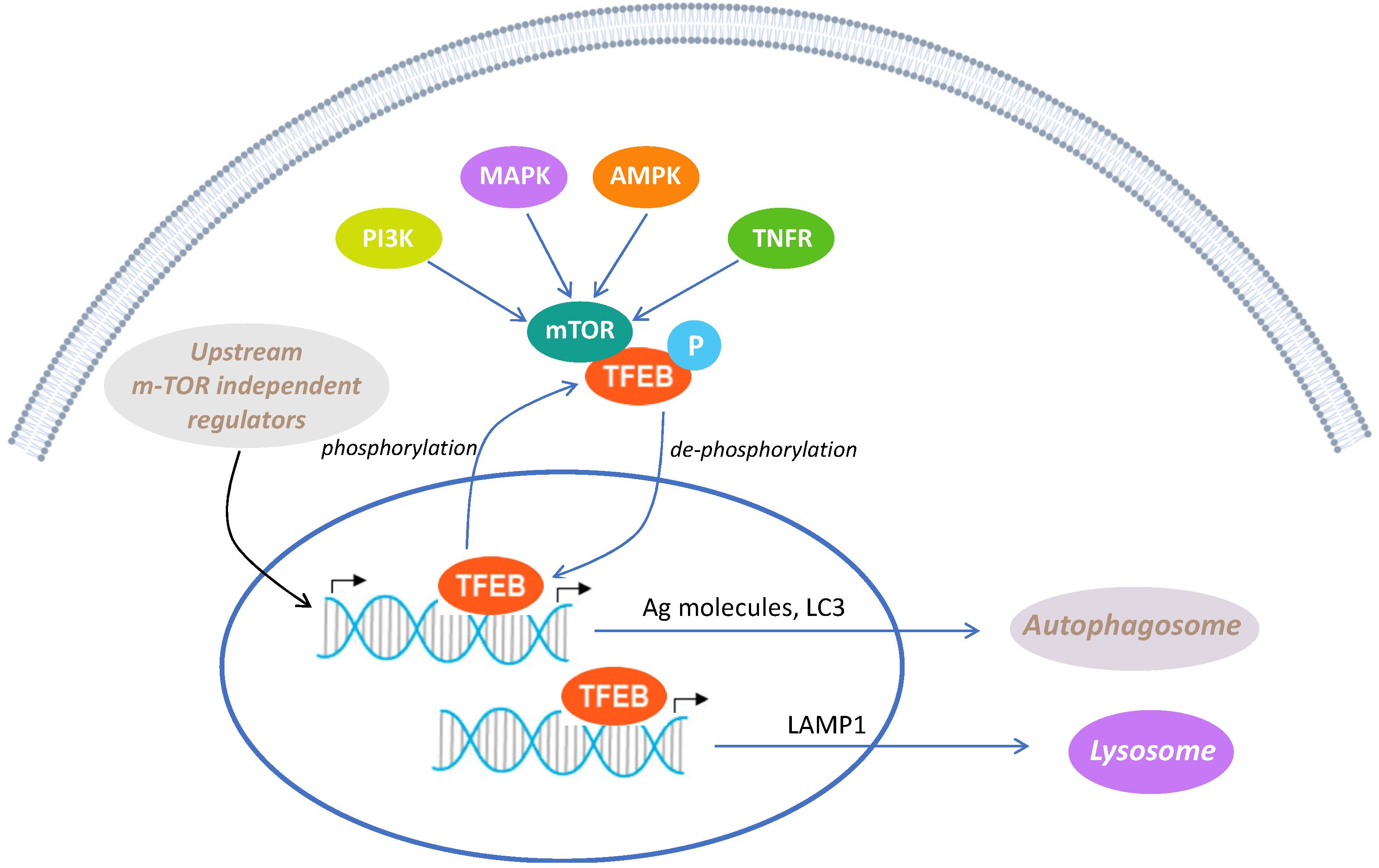
3.3. FD22a Prevents β-Amyloid-Induced Down-Regulation of Autophagy in U87-MG Cells
3.4. Quantitative Proteomic Analysis Uncovers the Activation of TFEB in the Restoration of Autophagy by FD22a
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Rubinsztein, D.C. Control of Autophagy as a Therapy for Neurodegenerative Disease. Nat. Rev. Neurol. 2012, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, J.; Gehl, C.R.; Long, J.D.; Johnson, H.; Magnotta, V.A.; Sewell, D.; Shannon, K.; Paulsen, J.S. Mild Cognitive Impairment as an Early Landmark in Huntington’s Disease. Front. Neurol. 2021, 12, 678652. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-Mediated Autophagy Rescues Midbrain Dopamine Neurons from α-Synuclein Toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, R.; Carrera, I.; Xu, S.; Lakshmana, M.K. TFEB Overexpression in the P301S Model of Tauopathy Mitigates Increased PHF1 Levels and Lipofuscin Puncta and Rescues Memory Deficits. eNeuro 2016, 3, ENEURO.0042-16.2016. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The Complex Relationship between TFEB Transcription Factor Phosphorylation and Subcellular Localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal Calcium Signalling Regulates Autophagy through Calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Cortes, C.J.; Miranda, H.C.; Frankowski, H.; Batlevi, Y.; Young, J.E.; Le, A.; Ivanov, N.; Sopher, B.L.; Carromeu, C.; Muotri, A.R.; et al. Polyglutamine-Expanded Androgen Receptor Interferes with TFEB to Elicit Autophagy Defects in SBMA. Nat. Neurosci. 2014, 17, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α Rescues Huntington’s Disease Proteotoxicity by Preventing Oxidative Stress and Promoting TFEB Function. Sci. Transl. Med. 2012, 4, 142ra97. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; Di Ronza, A.; Sardiello, M.; Segatori, L. TFEB Regulates Lysosomal Proteostasis. Human. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; Di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR Network Reveals an Integrated Control of Cellular Clearance Pathways. Human. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Ballabio, A. TFEB Regulates Autophagy: An Integrated Coordination of Cellular Degradation and Recycling Processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, R.; Laezza, C.; Bifulco, M.; Marasco, D.; Malfitano, A. Endocannabinoid System in Neurological Disorders. Recent Pat. CNS Drug Discov. 2016, 10, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Romero, J.; Ramos, J.A. Endocannabinoids and Neurodegenerative Disorders: Parkinson’s Disease, Huntington’s Chorea, Alzheimer’s Disease, and Others. In Endocannabinoids; Pertwee, R.G., Ed.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2015; Volume 231, pp. 233–259. ISBN 978-3-319-20824-4. [Google Scholar]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The Endocannabinoid System: An Overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua-Olaizola, O.; Elezgarai, I.; Rico-Barrio, I.; Zarandona, I.; Etxebarria, N.; Usobiaga, A. Targeting the Endocannabinoid System: Future Therapeutic Strategies. Drug Discov. Today 2017, 22, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.-C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Navarra, G.; Coppola, L.; Avilia, G.; Bifulco, M.; Laezza, C. Cannabinoids: Therapeutic Use in Clinical Practice. Int. J. Mol. Sci. 2022, 23, 3344. [Google Scholar] [CrossRef] [PubMed]
- Kibret, B.G.; Ishiguro, H.; Horiuchi, Y.; Onaivi, E.S. New Insights and Potential Therapeutic Targeting of CB2 Cannabinoid Receptors in CNS Disorders. Int. J. Mol. Sci. 2022, 23, 975. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Yu, W.; Ying, J.; Fang, P.; Zheng, Q.; Feng, X.; Hu, J.; Xiao, F.; Chen, S.; et al. CB2R Activation Regulates TFEB-Mediated Autophagy and Affects Lipid Metabolism and Inflammation of Astrocytes in POCD. Front. Immunol. 2022, 13, 836494. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Ferrisi, R.; Polini, B.; Mohamed, K.A.; Ricardi, C.; Lucarini, E.; Carpi, S.; Domenichini, F.; Stevenson, L.A.; Rapposelli, S.; et al. Design, Synthesis, and Biological Activity of New CB2 Receptor Ligands: From Orthosteric and Allosteric Modulators to Dualsteric/Bitopic Ligands. J. Med. Chem. 2022, 65, 9918–9938. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Sexton, P.M.; Christopoulos, A. Bridging the Gap: Bitopic Ligands of G-Protein-Coupled Receptors. Trends Pharmacol. Sci. 2013, 34, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Battiti, F.O.; Bonifazi, A. 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem. 2020, 63, 1779–1797. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and Biological Activities of Amyloid Beta Peptide 25–35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Polini, B.; Ricardi, C.; Bertolini, A.; Carnicelli, V.; Rutigliano, G.; Saponaro, F.; Zucchi, R.; Chiellini, G. T1AM/TAAR1 System Reduces Inflammatory Response and β-Amyloid Toxicity in Human Microglial HMC3 Cell Line. Int. J. Mol. Sci. 2023, 24, 11569. [Google Scholar] [CrossRef]
- Ciregia, F.; Bugliani, M.; Ronci, M.; Giusti, L.; Boldrini, C.; Mazzoni, M.R.; Mossuto, S.; Grano, F.; Cnop, M.; Marselli, L.; et al. Palmitate-Induced Lipotoxicity Alters Acetylation of Multiple Proteins in Clonal β Cells and Human Pancreatic Islets. Sci. Rep. 2017, 7, 13445. [Google Scholar] [CrossRef] [PubMed]
- Barbalace, M.C.; Zallocco, L.; Beghelli, D.; Ronci, M.; Scortichini, S.; Digiacomo, M.; Macchia, M.; Mazzoni, M.R.; Fiorini, D.; Lucacchini, A.; et al. Antioxidant and Neuroprotective Activity of Extra Virgin Olive Oil Extracts Obtained from Quercetano Cultivar Trees Grown in Different Areas of the Tuscany Region (Italy). Antioxidants 2021, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Lacerenza, S.; Ciregia, F.; Giusti, L.; Bonotti, A.; Greco, V.; Giannaccini, G.; D’Antongiovanni, V.; Fallahi, P.; Pieroni, L.; Cristaudo, A.; et al. Putative Biomarkers for Malignant Pleural Mesothelioma Suggested by Proteomic Analysis of Cell Secretome. Cancer Genom. Proteom. 2020, 17, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ciregia, F.; Giusti, L.; Da Valle, Y.; Donadio, E.; Consensi, A.; Giacomelli, C.; Sernissi, F.; Scarpellini, P.; Maggi, F.; Lucacchini, A.; et al. A Multidisciplinary Approach to Study a Couple of Monozygotic Twins Discordant for the Chronic Fatigue Syndrome: A Focus on Potential Salivary Biomarkers. J. Transl. Med. 2013, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Giusti, L.; Angeloni, C.; Barbalace, M.; Lacerenza, S.; Ciregia, F.; Ronci, M.; Urbani, A.; Manera, C.; Digiacomo, M.; Macchia, M.; et al. A Proteomic Approach to Uncover Neuroprotective Mechanisms of Oleocanthal against Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 2329. [Google Scholar] [CrossRef] [PubMed]
- Pontifex, M.G.; Connell, E.; Le Gall, G.; Pourtau, L.; Gaudout, D.; Angeloni, C.; Zallocco, L.; Ronci, M.; Giusti, L.; Müller, M.; et al. Saffron Extract (Safr’InsideTM) Improves Anxiety Related Behaviour in a Mouse Model of Low-Grade Inflammation through the Modulation of the Microbiota and Gut Derived Metabolites. Food Funct. 2022, 13, 12219–12233. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Liang, D.; Chen, Y.; Xie, Y.; Wang, Y.; Wang, L.; Wang, Z.; Qiao, Z. Gx-50 Reduces β-Amyloid-Induced TNF-α, IL-1β, NO, and PGE 2 Expression and Inhibits NF-κB Signaling in a Mouse Model of Alzheimer’s Disease. Eur. J. Immunol. 2016, 46, 665–676. [Google Scholar] [CrossRef]
- Cisbani, G.; Rivest, S. Targeting Innate Immunity to Protect and Cure Alzheimer’s Disease: Opportunities and Pitfalls. Mol. Psychiatry 2021, 26, 5504–5515. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Bian, J.-S. Hydrogen Sulfide Protects Amyloid-β Induced Cell Toxicity in Microglia. J. Alzheimer’s Dis. 2011, 22, 1189–1200. [Google Scholar] [CrossRef]
- Ranaivo, H.R.; Craft, J.M.; Hu, W.; Guo, L.; Wing, L.K.; Van Eldik, L.J.; Watterson, D.M. Glia as a Therapeutic Target: Selective Suppression of Human Amyloid-β-Induced Upregulation of Brain Proinflammatory Cytokine Production Attenuates Neurodegeneration. J. Neurosci. 2006, 26, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The Role of Autophagy in Neurodegenerative Disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. The mTOR Signalling Cascade: Paving New Roads to Cure Neurological Disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR Signaling Occurs Early in the Progression of Alzheimer Disease (AD): Analysis of Brain from Subjects with Pre-clinical AD, Amnestic Mild Cognitive Impairment and Late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Biagioni, F.; Antonietta Oliva, M.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin Inhibits the Growth of Glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Ryskalin, L.; Biagioni, F.; Gambardella, S.; Busceti, C.L.; Falleni, A.; Lazzeri, G.; Fornai, F. Methamphetamine Increases Prion Protein and Induces Dopamine-Dependent Expression of Protease Resistant PrPsc. Arch. Ital. Biol. 2017, 155, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The Emerging Role of M-TOR up-Regulation in Brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Bellusci, L.; Laurino, A.; Sabatini, M.; Sestito, S.; Lenzi, P.; Raimondi, L.; Rapposelli, S.; Biagioni, F.; Fornai, F.; Salvetti, A.; et al. New Insights into the Potential Roles of 3-Iodothyronamine (T1AM) and Newly Developed Thyronamine-Like TAAR1 Agonists in Neuroprotection. Front. Pharmacol. 2017, 8, 905. [Google Scholar] [CrossRef]
- Franco-Juárez, B.; Coronel-Cruz, C.; Hernández-Ochoa, B.; Gómez-Manzo, S.; Cárdenas-Rodríguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Ávila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. [Google Scholar] [CrossRef]
- Meyer, K.; Kaspar, B.K. Glia–Neuron Interactions in Neurological Diseases: Testing Non-Cell Autonomy in a Dish. Brain Res. 2017, 1656, 27–39. [Google Scholar] [CrossRef]
- Rahman, M.; Islam, R.; Yamin, M.; Islam, M.M.; Sarker, M.T.; Meem, A.F.K.; Akter, A.; Bin Emran, T.; Cavalu, S.; Sharma, R. Emerging Role of Neuron-Glia in Neurological Disorders: At a Glance. Oxidative Med. Cell. Longev. 2022, 2022, 3201644. [Google Scholar] [CrossRef] [PubMed]
- Diab, R.; Pilotto, F.; Saxena, S. Autophagy and Neurodegeneration: Unraveling the Role of C9ORF72 in the Regulation of Autophagy and Its Relationship to ALS-FTD Pathology. Front. Cell. Neurosci. 2023, 17, 1086895. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Álvarez, M.; Strippoli, R.; Donadelli, M.; Bazhin, A.V.; Cordani, M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers 2019, 11, 1415. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Song, Z.-H. Molecular and Cellular Changes Induced by the Activation of CB2 Cannabinoid Receptors in Trabecular Meshwork Cells. Mol. Vis. 2007, 13, 1348–1356. [Google Scholar] [PubMed]
- Yang, L.; Li, F.-F.; Han, Y.-C.; Jia, B.; Ding, Y. Cannabinoid Receptor CB2 Is Involved in Tetrahydrocannabinol-Induced Anti-Inflammation against Lipopolysaccharide in MG-63 Cells. Mediat. Inflamm. 2015, 2015, 362126. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 Functions as a Transcriptional Regulator of Autophagy by Preventing Nuclear Transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Peña-Llopis, S.; Konda, M.; Brugarolas, J. Multistep Regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Sequence (RefSeq) RNA | Gene Symbol | Primer Sequences |
|---|---|---|
| NM_002046 | GAPDH | (F) 5′-CCCTTCATTGACCTCAACTACATG |
| (R) 5′-TGGGATTTCCATTGATGACAAGC | ||
| NM_022818.5 | LC3 | (F) 5′-AGTCTTCTCTTCAGGTTCAC |
| (R) 5′-CTCACACAGCCCGTTTAC | ||
| NM_004958.4 | MTOR | (F) 5′-TGCCTTCACAGATACCCAG |
| (R) 5′-AGACCTCACAGCCACAGA | ||
| NM_001282208 | SIGMAR1 | (F) 5′-CTTCTACCCAGGGGAGAC |
| (R) 5′-GCATAGGAGCGAAGAGTAT | ||
| NM_001314049.2 | SIRT1 | (F) 5′-GGGTTCTTCTAAACTTGGACTCT |
| (R) 5′-GTAGGCGGCTTGATGGTAAT | ||
| NM_054354940.1 | SIRT5 | (F) 5′-CAAATCTGGTTTCGTGTGGAC |
| (R) 5′-AATAACTAAAGCCCGCCTCAA | ||
| NM_001193285 | SIRT6 | (F) 5′-CTCCTCCGCTTCCTGGTC |
| (R) 5′-TTACACTTGGCACATTCTTCC | ||
| NM_002982.4 | MCP1 | (F) 5′-GAGAGGCTGAGACTAACC |
| (R) 5′-TGATTGCATCTGGCTGAG | ||
| NM_001404662 | NFKB | (F) 5′-CCTTTCTCATCCCATCTTT |
| (R) 5′-CCTCAATGTCCTCTTTCTG |
| # | ID | Gene | Protein Name | Score | Cov | Pep | pI | MW | p-Value | Ratio FD22a + Aβ25–35/Aβ25–35 |
|---|---|---|---|---|---|---|---|---|---|---|
| 996 | P08238 | HS90B | Heat shock protein HSP 90-beta | 70 | 9 | 6 | 4.97 | 83,543 | 0.005 | 0.67 |
| 998 | P07900 | HS90A | Heat shock protein HSP 90-alpha | 72 * | 3 | 1 | 4.94 | 84,660 | 0.022 | 0.71 |
| 999 | P07900 | HS90A | Heat shock protein HSP 90-alpha | 73 * | 3 | 1 | 4.94 | 84,660 | 0.036 | 0.65 |
| 999 | P08238 | HS90B | Heat shock protein HSP 90-beta | 73 * | 3 | 1 | 4.96 | 83,264 | 0.036 | 0.65 |
| 1036 | Q92499 | DDX1 | ATP-dependent RNA helicase | 84 * | 5 | 4 | 6.8 | 82,432 | 0.076 | 1.29 |
| 1178 | P02545 | LMNA | Prelamin-A/C | 159 * | 18 | 12 | 6.57 | 74,140 | 0.005 | 1.22 |
| 1229 | P38646 | HSPA9 | Stress-70 protein. mitochondrial | 73 | 11 | 6 | 5.87 | 73,920 | 0.008 | 1.16 |
| 1288 | O94826 | TOM70 | Mitochondrial import receptor subunit TOM70 | 45 * | 5 | 2 | 6.82 | 67,455 | 0.038 | 0.79 |
| 1518 | P48643 | TCPE | T-complex protein 1 subunit epsilon | 40 * | 2 | 1 | 6.1 | 60,534 | 0.143 | 0.77 |
| 1645 | Q13153 | PAK1 | Serine/threonine-protein kinase PAK 1 | 59 | 19 | 7 | 5.55 | 60,894 | 0.023 | 1.35 |
| 1730 | P78371 | CCT2 | T-complex protein 1 subunit beta | 150 | 32 | 11 | 6.01 | 57,794 | 0.000 | 1.14 |
| 1769 | Q07960 | RHG01 | Rho GTPase-activating protein 1 | 36 * | 3 | 1 | 5.85 | 50,436 | 0.249 | 0.83 |
| 2101 | Q15019 | SEPTIN2 | Septin-2 | 64 | 18 | 4 | 6.15 | 41,689 | 0.023 | 1.21 |
| 2200 | P04075 | ALDOA | Fructose-bisphosphate aldolase A | 139 * | 9 | 9 | 8.39 | 39,420 | 0.001 | 1.26 |
| 2396 | P04083 | ANXA1 | Annexin A1 | 174 | 39 | 11 | 6.57 | 38,918 | 0.004 | 0.92 |
| 2896 | Q9HB71 | CYBP | Calcyclin-binding protein | 59 * | 7 | 2 | 8.32 | 26,210 | 0.005 | 0.79 |
| 2956 | P04792 | HSPB1 | Heat shock protein beta-1 | 66 | 16 | 4 | 5.98 | 22,826 | 0.006 | 0.78 |
| 2973 | P52565 | ARHGDIA | Rho GDP-dissociation inhibitor 1 | 78 | 29 | 6 | 5.02 | 23,250 | 0.009 | 1.16 |
| 3025 | P09936 | UCHL1 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 56 | 19 | 3 | 5.33 | 25,151 | 0.001 | 1.18 |
| 3269 | P32119 | PRDX2 | Peroxiredoxin-2 | 70 | 22 | 4 | 5.66 | 22,049 | 0.084 | 1.20 |
| 3944 | P23528 | CFL1 | Cofilin-1 | 120 | 48 | 7 | 8.22 | 18,719 | 0.001 | 0.63 |
| # | ID | Gene | Protein Name | Score | Cov | Pep | pI | MW | p-Value | Ratio FD22a/Ctrl |
|---|---|---|---|---|---|---|---|---|---|---|
| 858 | P12814 | ACTN1 | Alpha-actinin-1 | 82 | 11 | 8 | 5.25 | 103,563 | 0.0107 | 0.70 |
| 914 | P55072 | VCP | Transitional endoplasmic reticulum ATPase | 190 | 28 | 18 | 5.14 | 89,950 | 0.0478 | 0.69 |
| 1140 | Q9NTI5 | PDS5B | Sister chromatid cohesion protein PDS5 homolog B | 56 | 7 | 9 | 8.67 | 16,5818 | 0.0129 | 0.67 |
| 1161 | P11021 | HSPA5 | Endoplasmic reticulum chaperone BiP | 237 | 36 | 18 | 5.07 | 72,402 | 0.0035 | 0.68 |
| 1199 | P41250 | GARS1 | Glycine--tRNA ligase | 116 | 21 | 10 | 6.61 | 83,854 | 0.0010 | 1.37 |
| 1205 | P26038 | MSN | Moesin | 87 | 14 | 8 | 6.08 | 67,892 | 0.0242 | 1.48 |
| 1278 | P11142 | HSPA8 | Heat shock cognate 71 kDa protein | 89 | 14 | 7 | 5.37 | 71,082 | 0.0273 | 1.28 |
| 1301 | P11142 | HSPA8 | Heat shock cognate 71 kDa protein | 55 | 9 | 5 | 5.37 | 71,082 | 0.0184 | 1.35 |
| 1317 | O75864 | PPP1R37 | Protein phosphatase 1 regulatory subunit 37 | 47 | 6 | 3 | 4.97 | 74,767 | 0.0435 | 0.83 |
| 1421 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 78 | 20 | 7 | 5.39 | 51,230 | 0.0017 | 1.44 |
| 1432 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 150 | 25 | 10 | 5.39 | 51,230 | 0.0457 | 1.26 |
| 1435 | Q9H6N6 | MYH16 | Putative uncharacterized protein MYH16 | 64 | 14 | 12 | 5.4 | 128,439 | 0.0051 | 0.76 |
| 1443 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 96 | 23 | 8 | 5.39 | 51,230 | 0.0069 | 1.33 |
| 1451 | Q16555 | DPYSL2 | Dihydropyrimidinase-related protein 2 | 58 | 14 | 5 | 5.95 | 62,711 | 0.0014 | 0.77 |
| 1461 | P48643 | TCPG | T-complex protein 1 subunit gamma | 93 * | 4 | 2 | 6.1 | 60,534 | 0.0076 | 0.82 |
| 1461 | Q16555 | DPYL2 | Dihydropyrimidinase-related protein 2 | 54 * | 2 | 1 | 5.95 | 62,294 | 0.0076 | 0.82 |
| 1511 | P08670 | VIM | Vimentin | 147 | 39 | 14 | 5.06 | 53,676 | 0.0274 | 0.78 |
| 1524 | P10809 | HSPD1 | 60 kDa heat shock protein. mitochondrial | 203 | 41 | 16 | 5.7 | 61,187 | 0.0308 | 1.17 |
| 1610 | P08670 | VIM | Vimentin | 221 | 48 | 21 | 5.06 | 53,676 | 0.0156 | 1.20 |
| 1641 | P68371 | TUBB4B | Tubulin beta-4B chain | 142 | 31 | 13 | 4.79 | 50,255 | 0.0043 | 0.51 |
| 1725 | O43175 | PHGDH | D-3-phosphoglycerate dehydrogenase | 56 | 12 | 6 | 6.29 | 57,356 | 0.0005 | 1.81 |
| 1726 | P28329 | CHAT | Choline O-acetyltransferase | 73 | 9 | 8 | 8.9 | 83,852 | 0.0282 | 0.74 |
| 1730 | P78371 | CCT2 | T-complex protein 1 subunit beta | 150 | 32 | 11 | 6.01 | 57,794 | 0.0271 | 0.88 |
| 1745 | O00148 | DDX39A | ATP-dependent RNA helicase | 92 | 19 | 8 | 5.46 | 49,611 | 0.0263 | 0.73 |
| 1848 | P48594 | SERPINB4 | Serpin B4 | 60 | 12 | 5 | 5.86 | 44,997 | 0.0192 | 1.20 |
| 1859 | P06733 | ENO1 | Alpha-enolase | 61 | 17 | 5 | 7.01 | 47,481 | 0.0479 | 1.29 |
| 1864 | P08670 | VIM | Vimentin | 70 | 29 | 12 | 5.06 | 53,676 | 0.0056 | 0.48 |
| 1877 | P31930 | UQCRC1 | Cytochrome b-c1 complex subunit 1. mitochondrial | 84 | 27 | 7 | 5.94 | 53,297 | 0.0397 | 1.20 |
| 1951 | P08670 | VIM | Vimentin | 123 | 24 | 10 | 5.06 | 53,676 | 0.0117 | 0.40 |
| 2042 | O75874 | IDHC | Isocitrate dehydrogenase [NADP] cytoplasmic (IDH) | 124 * | 17 | 7 | 6.53 | 45,659 | 0.0491 | 0.85 |
| 2077 | Q7L2H7 | EIF3M | Eukaryotic translation initiation factor 3 subunit M | 62 | 15 | 4 | 5.41 | 42,932 | 0.0091 | 1.51 |
| 2140 | Q9UNM6 | PSMD13 | 26S proteasome non-ATPase regulatory subunit 13 | 86 | 18 | 5 | 5.53 | 43,203 | 0.0492 | 1.16 |
| 2365 | P67775 | PPP2CA | Serine/threonine-protein phosphatase 2A catalytic subunit alpha isoform | 66 | 17 | 4 | 5.3 | 36,142 | 0.0425 | 1.24 |
| 2464 | P31942 | HNRNPH3 | Heterogeneous nuclear ribonucleoprotein H3 | 70 | 19 | 5 | 6.37 | 36,960 | 0.0228 | 1.36 |
| 2491 | P62879 | GNB2 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | 71 | 18 | 6 | 5.6 | 38,048 | 0.0029 | 1.43 |
| 2595 | P09525 | ANXA4 | Annexin A4 | 59 | 15 | 4 | 5.84 | 36,088 | 0.0169 | 1.32 |
| 2637 | P07951 | TPM2 | Tropomyosin beta chain | 62 | 26 | 7 | 4.66 | 32,945 | 0.0171 | 1.49 |
| 2653 | P06753 | TPM3 | Tropomyosin alpha 3 chain | 70 | 13 | 6 | 4.68 | 32,987 | 0.0001 | 0.68 |
| 2720 | P62258 | YWHAE | 14-3-3 protein epsilon | 79 | 31 | 6 | 4.63 | 29,326 | 0.0014 | 0.54 |
| 2806 | P61981 | YWHAG | 14-3-3 protein gamma | 68 | 27 | 5 | 4.8 | 28,456 | 0.0000 | 0.55 |
| 2864 | P63104 | YWHAZ | 14-3-3 protein zeta/delta | 87 | 31 | 6 | 4.73 | 27,899 | 0.0001 | 0.39 |
| 2875 | P17480 | UBTF | Nucleolar transcription factor 1 | 59 | 10 | 8 | 5.63 | 89,692 | 0.0221 | 1.34 |
| 2884 | P31946 | YWHAB | 14-3-3 protein beta/alpha | 88 | 27 | 6 | 4.76 | 28,179 | 0.0009 | 0.65 |
| 2895 | P04083 | ANXA1 | Annexin A1 | 65 | 16 | 4 | 6.57 | 38,918 | 0.0012 | 1.75 |
| 2932 | O75489 | NDUFS3 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 3. mitochondrial | 74 | 24 | 5 | 6.99 | 30,337 | 0.0116 | 1.28 |
| 2997 | P30041 | PRDX6 | Peroxiredoxin-6 | 94 | 26 | 5 | 6 | 25,133 | 0.0046 | 0.81 |
| 4526 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 95 | 23 | 8 | 5.39 | 51,230 | 0.0314 | 0.76 |
| # | ID | Gene | Protein Name | Score | Cov | Pep | pI | MW | p-Value | Ratio Aβ25–35/Ctrl |
|---|---|---|---|---|---|---|---|---|---|---|
| 669 | P53396 | ACLY | ATP-citrate synthase | 72 * | 2 | 2 | 6.95 | 120,839 | 0.0151 | 0.76 |
| 669 | P53992 | SC24C | Protein transport protein Sec24C | 63 * | 2 | 2 | 6.71 | 118,325 | 0.0151 | 0.76 |
| 1099 | P13798 | APEH | Acylamino-acid-releasing enzyme | 58 | 12 | 7 | 5.29 | 82,142 | 0.0485 | 1.29 |
| 1136 | P0CG48 | UBC | Polyubiquitin-C | 49 * | 2 | 1 | 7.16 | 77,039 | 0.0007 | 0.43 |
| 1140 | Q9NTI5 | PDS5B | Sister chromatid cohesion protein PDS5 homolog B | 56 | 7 | 9 | 8.67 | 165,818 | 0.0036 | 0.43 |
| 1149 | P02545 | LMNA | Prelamin-A/C [Cleaved into: Lamin-A/C (70 kDa lamin) (Renal carcinoma antigen NY-REN-32)] | 41 * | 2 | 1 | 6.57 | 74,140 | 0.0185 | 0.55 |
| 1178 | P02545 | LMNA | Prelamin-A/C | 159 * | 18 | 12 | 6.57 | 74,140 | 0.0051 | 0.62 |
| 1179 | P41250 | GARS | Glycine--tRNA ligase | 79 * | 3 | 3 | 6.61 | 83,166 | 0.0144 | 1.39 |
| 1314 | P20700 | LMNB1 | Lamin-B1 | 80 | 11 | 8 | 5.11 | 66,653 | 0.0330 | 0.74 |
| 1317 | O75864 | PPP1R37 | Protein phosphatase 1 regulatory subunit 37 | 47 | 6 | 3 | 4.97 | 74,767 | 0.0078 | 0.58 |
| 1331 | Q9NSD9 | SYFB | Phenylalanine--tRNA ligase beta subunit | 90 * | 8 | 8 | 6.39 | 66,116 | 0.0524 | 0.65 |
| 1356 | P31040 | SDHA | Succinate dehydrogenase [ubiquinone] flavoprotein subunit mitochondrial | 94 * | 4 | 4 | 7.06 | 72,692 | 0.0021 | 0.54 |
| 1432 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 150 | 25 | 10 | 5.39 | 51,230 | 0.1540 | 1.36 |
| 1451 | Q16555 | DPYSL2 | Dihydropyrimidinase-related protein 2 | 58 | 14 | 5 | 5.95 | 62,711 | 0.0002 | 0.86 |
| 1461 | P48643 | TCPG | T-complex protein 1 subunit gamma | 93 * | 4 | 2 | 6.1 | 60,534 | 0.0002 | 0.87 |
| 1461 | Q16555 | DPYL2 | Dihydropyrimidinase-related protein 2 | 54 * | 2 | 1 | 5.95 | 62,294 | 0.0002 | 0.87 |
| 2042 | O75874 | IDHC | Isocitrate dehydrogenase [NADP] cytoplasmic | 124 * | 17 | 7 | 6.53 | 45,659 | 0.0028 | 0.86 |
| 2200 | P04075 | ALDOA | Fructose-bisphosphate aldolase A | 139 * | 9 | 9 | 8.39 | 39,420 | 0.0015 | 0.77 |
| 2896 | Q9HB71 | CYBP | Calcyclin-binding protein | 59 * | 7 | 2 | 8.32 | 26,210 | 0.0055 | 1.32 |
| 2997 | P30041 | PRDX6 | Peroxiredoxin-6 | 94 | 26 | 5 | 6 | 25,133 | 0.0002 | 0.81 |
| 3944 | P23528 | CFL1 | Cofilin-1 | 120 | 48 | 7 | 8.22 | 18,719 | 0.0009 | 0.75 |
| 4526 | P61978 | HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 95 | 23 | 8 | 5.39 | 51,230 | 0.0008 | 0.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polini, B.; Zallocco, L.; Gado, F.; Ferrisi, R.; Ricardi, C.; Zuccarini, M.; Carnicelli, V.; Manera, C.; Ronci, M.; Lucacchini, A.; et al. A Proteomic Approach Identified TFEB as a Key Player in the Protective Action of Novel CB2R Bitopic Ligand FD22a against the Deleterious Effects Induced by β-Amyloid in Glial Cells. Cells 2024, 13, 875. https://doi.org/10.3390/cells13100875
Polini B, Zallocco L, Gado F, Ferrisi R, Ricardi C, Zuccarini M, Carnicelli V, Manera C, Ronci M, Lucacchini A, et al. A Proteomic Approach Identified TFEB as a Key Player in the Protective Action of Novel CB2R Bitopic Ligand FD22a against the Deleterious Effects Induced by β-Amyloid in Glial Cells. Cells. 2024; 13(10):875. https://doi.org/10.3390/cells13100875
Chicago/Turabian StylePolini, Beatrice, Lorenzo Zallocco, Francesca Gado, Rebecca Ferrisi, Caterina Ricardi, Mariachiara Zuccarini, Vittoria Carnicelli, Clementina Manera, Maurizio Ronci, Antonio Lucacchini, and et al. 2024. "A Proteomic Approach Identified TFEB as a Key Player in the Protective Action of Novel CB2R Bitopic Ligand FD22a against the Deleterious Effects Induced by β-Amyloid in Glial Cells" Cells 13, no. 10: 875. https://doi.org/10.3390/cells13100875