



Ezetimibe Induces Paraptosis through Niemann–Pick C1-like 1 Inhibition of Mammalian-Target-of-Rapamycin Signaling in Hepatocellular Carcinoma Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Plasmid Transfection
2.3. Measurement of Cell Viability
2.4. Colony Formation Assay
2.5. Microscopy, Electron Microscopy, and Confocal Laser Scanning Microscopy
2.6. EdU Incorporation Assay
2.7. 20S Proteasome Activity Assay
2.8. Apoptosis Analysis
2.9. Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR)
2.10. Western Blot Analysis
2.11. In Vivo Tumor Xenograft Studies
2.12. Statistical Analyses
3. Results
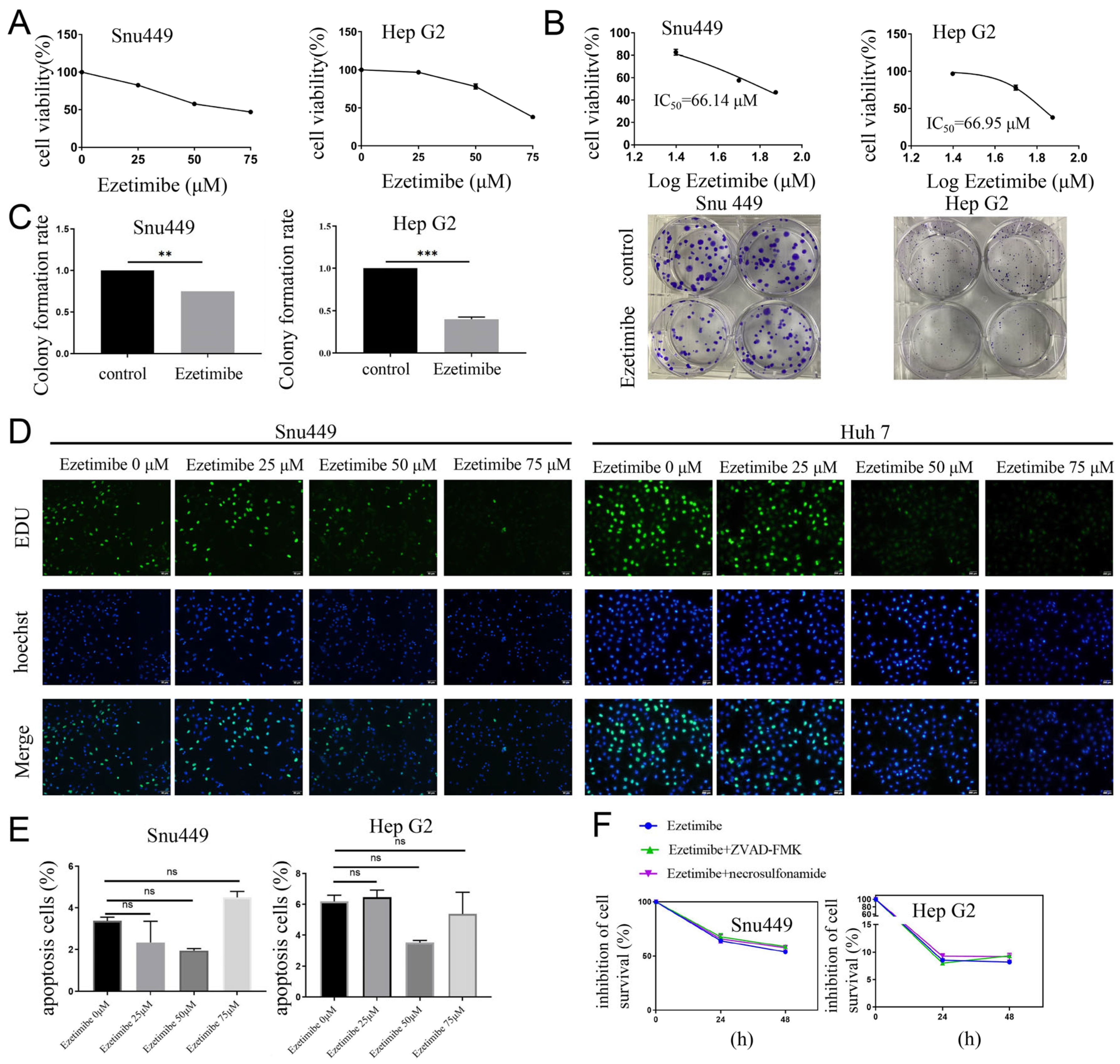
3.1. Ezetimibe Treatment Induces Cell Death That Is Independent of Apoptosis
3.2. Ezetimibe Produces Massive Cytoplasmic Vacuolation in HCC Cells
3.3. Ezetimibe-Induced Cell Vacuolization Originates from ER
3.4. Ezetimibe-Induced ER Stress and ROS Generation Result in Paraptosis in HCC Cells
3.5. Ezetimibe Induces Paraptosis by Inhibiting Proteasome Activity
3.6. Ezetimibe Induces Paraptosis by Targeting NPC1L1 to Enhance MAPK Pathway and Inhibit mTOR Pathway in HCC Cells
3.7. Ezetimibe Inhibits Growth of Hepatocellular Carcinoma Tumor in Xenograft Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Liu, X.; Gu, Y.; Bian, Y.; Cai, D.; Li, Y.; Zhao, Y.; Zhang, Z.; Xue, M.; Zhang, L. Honokiol induces paraptosis-like cell death of acute promyelocytic leukemia via mTOR & MAPK signaling pathways activation. Apoptosis 2021, 26, 195–208. [Google Scholar] [CrossRef]
- Liu, T.; Xie, X.L.; Zhou, X.; Chen, S.X.; Wang, Y.J.; Shi, L.P.; Chen, S.J.; Wang, Y.J.; Wang, S.L.; Zhang, J.N.; et al. Y-box binding protein 1 augments sorafenib resistance via the PI3K/Akt signaling pathway in hepatocellular carcinoma. World J. Gastroenterol. 2021, 27, 4667–4686. [Google Scholar] [CrossRef]
- Monel, B.; Compton, A.A.; Bruel, T.; Amraoui, S.; Burlaud-Gaillard, J.; Roy, N.; Guivel-Benhassine, F.; Porrot, F.; Genin, P.; Meertens, L.; et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. Embo J. 2017, 36, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 Forms an Alternative mTOR Complex with DNA-PKcs in Human Cancer. Iscience 2019, 17, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, o5838. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta-Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef]
- Valamanesh, F.; Torriglia, A.; Savoldelli, M.; Gandolphe, C.; Jeanny, J.C.; Benezra, D.; Behar-Cohen, F. Glucocorticoids induce retinal toxicity through mechanisms mainly associated with paraptosis. Mol. Vis. 2007, 13, 1746–1757. [Google Scholar]
- Wang, Y.; Xu, K.; Zhang, H.; Zhao, J.; Zhu, X.; Wang, Y.; Wu, R. Retinal ganglion cell death is triggered by paraptosis via reactive oxygen species production: A brief literature review presenting a novel hypothesis in glaucoma pathology. Mol. Med. Rep. 2014, 10, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, F.; Yang, Y.; Wang, P.; Zhang, Z.; Tang, Y.; Shen, Y.; Wang, K. Paraptosis-Inducing Nanomedicine Overcomes Cancer Drug Resistance for a Potent Cancer Therapy. Small 2018, 14, 1702446. [Google Scholar] [CrossRef] [PubMed]
- Ram, B.M.; Ramakrishna, G. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim. Biophys. Acta 2014, 1843, 2497–2512. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to delta-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, D.M.; Seo, M.J.; Lee, H.J.; Choi, K.S. Intracellular Ca(2+) Imbalance Critically Contributes to Paraptosis. Front. Cell. Dev. Biol. 2020, 8, 607844. [Google Scholar] [CrossRef]
- Altmann, S.W.; Davis, H.J.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef]
- Zhan, S.; Tang, M.; Liu, F.; Xia, P.; Shu, M.; Wu, X. Ezetimibe for the prevention of cardiovascular disease and all-cause mortality events. Cochrane Database Syst. Rev. 2018, 11, D12502. [Google Scholar] [CrossRef]
- Zhang, Z.; Qin, S.; Chen, Y.; Zhou, L.; Yang, M.; Tang, Y.; Zuo, J.; Zhang, J.; Mizokami, A.; Nice, E.C.; et al. Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. Embo Mol. Med. 2022, 14, e14903. [Google Scholar] [CrossRef]
- Schefe, J.H.; Lehmann, K.E.; Buschmann, I.R.; Unger, T.; Funke-Kaiser, H. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’s CT difference” formula. J. Mol. Med. 2006, 84, 901–910. [Google Scholar] [CrossRef]
- Lee, D.; Kim, I.Y.; Saha, S.; Choi, K.S. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol. Ther. 2016, 162, 120–133. [Google Scholar] [CrossRef]
- Pang, H.F.; Li, X.X.; Zhao, Y.H.; Kang, J.K.; Li, J.Y.; Tian, W.; Wang, C.M.; Hou, H.X.; Li, D.R. Confirming whether novel rhein derivative 4a induces paraptosis-like cell death by endoplasmic reticulum stress in ovarian cancer cells. Eur. J. Pharmacol. 2020, 886, 173526. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.J.; Lee, D.M.; Kim, I.Y.; Lee, D.; Choi, M.K.; Lee, J.Y.; Park, S.S.; Jeong, S.Y.; Choi, E.K.; Choi, K.S. Gambogic acid triggers vacuolization-associated cell death in cancer cells via disruption of thiol proteostasis. Cell Death Dis. 2019, 10, 187. [Google Scholar] [CrossRef]
- Lee, D.M.; Kim, I.Y.; Seo, M.J.; Kwon, M.R.; Choi, K.S. Nutlin-3 enhances the bortezomib sensitivity of p53-defective cancer cells by inducing paraptosis. Exp. Mol. Med. 2017, 49, e365. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Cui, M. Glabridin induces paraptosis-like cell death via ER stress in breast cancer cells. Heliyon 2022, 8, e10607. [Google Scholar] [CrossRef]
- Lai, K.M.; Wang, J.H.; Lin, S.C.; Wen, Y.; Wu, C.L.; Su, J.H.; Chen, C.C.; Lin, C.C. Crassolide Induces G2/M Cell Cycle Arrest, Apoptosis, and Autophagy in Human Lung Cancer Cells via ROS-Mediated ER Stress Pathways. Int. J. Mol. Sci. 2022, 23, 5624. [Google Scholar] [CrossRef]
- Pyrczak-Felczykowska, A.; Reekie, T.A.; Jakalski, M.; Hac, A.; Malinowska, M.; Pawlik, A.; Rys, K.; Guzow-Krzeminska, B.; Herman-Antosiewicz, A. The Isoxazole Derivative of Usnic Acid Induces an ER Stress Response in Breast Cancer Cells That Leads to Paraptosis-like Cell Death. Int. J. Mol. Sci. 2022, 23, 1802. [Google Scholar] [CrossRef]
- Kim, H.; Xue, X. Detection of Total Reactive Oxygen Species in Adherent Cells by 2′,7′-Dichlorodihydrofluorescein Diacetate Staining. J. Vis. Exp. 2020, e60682. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Dai, C.H.; Zhu, L.R.; Wang, Y.; Tang, X.P.; Du, Y.J.; Chen, Y.C.; Li, J. Celastrol acts synergistically with afatinib to suppress non-small cell lung cancer cell proliferation by inducing paraptosis. J. Cell. Physiol. 2021, 236, 4538–4554. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 215r–221r. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Li, W.; Diao, H.J.; Fan, R.Z.; Huang, J.L.; Gan, L.; Zou, M.F.; Tang, G.H.; Yin, S. Jolkinolide B targets thioredoxin and glutathione systems to induce ROS-mediated paraptosis and apoptosis in bladder cancer cells. Cancer Lett. 2021, 509, 13–25. [Google Scholar] [CrossRef]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Liebes, L.; Zou, Y.; Perez-Soler, R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 2003, 278, 33714–33723. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Chi, K.H.; Lin, W.W. Proteasome inhibitors stimulate activator protein-1 pathway via reactive oxygen species production. FEBS Lett. 2002, 526, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, S.; Calvaruso, G.; Lauricella, M.; Giuliano, M.; Bellavia, G.; D’Anneo, A.; Vento, R.; Tesoriere, G. Apoptosis induced in hepatoblastoma HepG2 cells by the proteasome inhibitor MG132 is associated with hydrogen peroxide production, expression of Bcl-XS and activation of caspase-3. Int. J. Oncol. 2002, 21, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, C.; Qin, X.; Liu, W.; Li, D.; Jia, H.; Gao, X.; Wu, Y.; Wu, Q.; Xu, X.; et al. DHW-221, a Dual PI3K/mTOR Inhibitor, Overcomes Multidrug Resistance by Targeting P-Glycoprotein (P-gp/ABCB1) and Akt-Mediated FOXO3a Nuclear Translocation in Non-small Cell Lung Cancer. Front. Oncol. 2022, 12, 873649. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.; Yang, C.; Huang, R.; Wen, Y.; Zhang, C.; Wu, C.; Zhao, B. Swainsonine Triggers Paraptosis via ER Stress and MAPK Signaling Pathway in Rat Primary Renal Tubular Epithelial Cells. Front. Pharmacol. 2021, 12, 715285. [Google Scholar] [CrossRef]
- Teymouri, M.; Barati, N.; Pirro, M.; Sahebkar, A. Biological and pharmacological evaluation of dimethoxycurcumin: A metabolically stable curcumin analogue with a promising therapeutic potential. J. Cell. Physiol. 2018, 233, 124–140. [Google Scholar] [CrossRef]
- Sperandio, S.; Poksay, K.; de Belle, I.; Lafuente, M.J.; Liu, B.; Nasir, J.; Bredesen, D.E. Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. [Google Scholar] [CrossRef]
- Bays, H. Ezetimibe. Expert Opin. Investig. Drugs 2002, 11, 1587–1604. [Google Scholar] [CrossRef]
- Park, S.W. Intestinal and hepatic niemann-pick c1-like 1. Diabetes Metab. J. 2013, 37, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Baraz, R.; Cisterne, A.; Saunders, P.O.; Hewson, J.; Thien, M.; Weiss, J.; Basnett, J.; Bradstock, K.F.; Bendall, L.J. mTOR inhibition by everolimus in childhood acute lymphoblastic leukemia induces caspase-independent cell death. PLoS ONE 2014, 9, e102494. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef]
- Sonntag, R.; Gassler, N.; Bangen, J.M.; Trautwein, C.; Liedtke, C. Pro-apoptotic Sorafenib signaling in murine hepatocytes depends on malignancy and is associated with PUMA expression in vitro and in vivo. Cell Death Dis. 2014, 5, e1030. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shu, C.; Laurence, A.D.; Chen, Y.; Peng, B.G.; Zhen, Z.J.; Cai, J.Q.; Ding, Y.T.; Li, L.Q.; Zhang, Y.B.; et al. Effect of Huaier granule on recurrence after curative resection of HCC: A multicentre, randomised clinical trial. Gut 2018, 67, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kim, E.H.; Kwon, T.K.; Park, S.A.; Choi, K.S. Simultaneous mitochondrial Ca(2+) overload and proteasomal inhibition are responsible for the induction of paraptosis in malignant breast cancer cells. Cancer Lett. 2012, 324, 197–209. [Google Scholar] [CrossRef]
- Cao, W.; Liu, X.; Zhang, Y.; Li, A.; Xie, Y.; Zhou, S.; Song, L.; Xu, R.; Ma, Y.; Cai, S.; et al. BEZ235 Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib by Inhibiting PI3K/AKT/mTOR and Inducing Autophagy. Biomed Res. Int. 2021, 2021, 5556306. [Google Scholar] [CrossRef]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.; Wu, C.; Zhou, Y.; Zhang, M.; Mai, S.; Chen, M.; Wang, H.-Y. Ezetimibe Induces Paraptosis through Niemann–Pick C1-like 1 Inhibition of Mammalian-Target-of-Rapamycin Signaling in Hepatocellular Carcinoma Cells. Genes 2024, 15, 4. https://doi.org/10.3390/genes15010004
Yin Y, Wu C, Zhou Y, Zhang M, Mai S, Chen M, Wang H-Y. Ezetimibe Induces Paraptosis through Niemann–Pick C1-like 1 Inhibition of Mammalian-Target-of-Rapamycin Signaling in Hepatocellular Carcinoma Cells. Genes. 2024; 15(1):4. https://doi.org/10.3390/genes15010004
Chicago/Turabian StyleYin, Yuting, Chun Wu, Yufeng Zhou, Meiyin Zhang, Shijuan Mai, Minshan Chen, and Hui-Yun Wang. 2024. "Ezetimibe Induces Paraptosis through Niemann–Pick C1-like 1 Inhibition of Mammalian-Target-of-Rapamycin Signaling in Hepatocellular Carcinoma Cells" Genes 15, no. 1: 4. https://doi.org/10.3390/genes15010004
APA StyleYin, Y., Wu, C., Zhou, Y., Zhang, M., Mai, S., Chen, M., & Wang, H.-Y. (2024). Ezetimibe Induces Paraptosis through Niemann–Pick C1-like 1 Inhibition of Mammalian-Target-of-Rapamycin Signaling in Hepatocellular Carcinoma Cells. Genes, 15(1), 4. https://doi.org/10.3390/genes15010004