
Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products


Institut de Combustion, Aérothermique, Réactivité et Environnement (ICARE), CNRS, 45071 Orléans, France
Atmosphere 2024, 15(5), 576; https://doi.org/10.3390/atmos15050576
Submission received: 23 April 2024
/
Revised: 3 May 2024
/
Accepted: 5 May 2024
/
Published: 8 May 2024
(This article belongs to the Special Issue Reaction Mechanisms and Chemical Kinetics in Atmospheric Chemistry)
Abstract
:Reaction OH + OCS → products (1) has been studied in a discharge–flow reactor combined with modulated molecular beam mass spectrometry. The reaction rate constant has been determined under pseudo-first-order conditions through monitoring OH decays in a high excess of OCS: k1 = (2.35 ± 0.25) × 10−12 exp(−(2144 ± 56)/T) cm3 molecule−1 s−1 at T = 365–960 K (the uncertainties represent precision at the 2σ level, the total 2σ relative uncertainty including statistical and systematic errors on the rate constant being 20% at all temperatures). The rate constant of reaction (1) was found to be similar at a total helium pressure of 1, 2, and 8 torr at around 500 K. The SH radical was identified as the primary product of the reaction, and its yield was determined to be about 100% at T = 500 and 750 K. The kinetic and mechanistic data from the present study are compared to previous experimental and theoretical work.
1. Introduction
Carbonyl sulfide (OCS) is the most abundant sulfur-containing gas phase compound in the troposphere. The ocean is thought to be the principal source of atmospheric OCS through direct emissions of OCS and oxidation of CS2 and DMS in the troposphere, along with anthropogenic sources, which have a comparable strength [1]. Uptake by vegetation and soil is the most important terrestrial sink of OCS [1]. The title reaction, although slow and thought to have a limited impact on the atmospheric budget of OCS,
it nevertheless represents the major gas-phase chemical process for OCS removal in the troposphere.
OH + OCS → products
In the past, the reaction has been studied many times, both experimentally [2,3,4,5,6,7,8] and theoretically [8,9,10]. However, many questions remain open, in particular the dependence of the reaction rate constant on pressure, temperature, and reaction products. Theoretical studies of the reaction are in clear contradiction with the experiment. They predict rate constants that are orders of magnitude lower than those measured experimentally, as well as the pressure dependence of the rate constant that has never been observed. Information on the reaction products, adduct vs. SH + CO2-forming channels, is also a subject of discrepancies between theory and experiment.
The present study aims to clarify at least some of these issues through experimental measurements of the reaction rate constant, including its temperature dependence over a wide temperature range (T = 365–960 K), pressure dependence between 1 and 8 torr total pressure of helium, and quantification of the yield of the SH radical, which was identified as the primary reaction product. The reaction kinetics at elevated temperatures, studied for the first time in this work, are of interest for modeling the chemistry of combustion and industrial processes [11], as well as the chemistry of hot near-source volcanic plumes [12].
2. Materials and Methods
Experiments were carried out in a standard-design discharge flow reactor combined with a mass spectrometric analysis (quadrupole mass spectrometer Balzers, QMG 420 with electron impact ionization) of the chemical composition of the reactive system [13,14]. The reactor consisted of a quartz tube (45 cm in length, 2.5 cm i.d.), where the temperature was controlled with electrical heating elements (Figure 1). Helium was used as a carrier gas in all the experiments, and the pumping speed was adjusted to produce linear flow velocities of 500–1670 cm s−1 in the pressure range of 1–8 Torr of the study.
Hydroxyl radicals were generated in the movable injector through the reaction
k2 = (1.47 ± 0.26) × 10−10 cm3 molecule−1 s−1 (T = 195–2000) [15], H atoms being produced by dissociation of H2, diluted in He, in a microwave discharge (Figure 1). In the experiments, where an NO2-free system was needed (product study), OH radicals were formed in a rapid reaction of F atoms with excess H2O (Figure 2):
k3 = (1.40 ± 0.15) × 10−11 cm3 molecule−1 s−1 (T = 240–380) [16]. Fluorine atoms were produced in the microwave discharge of trace amounts of F2 in He in an alumina tube (more than 95% of F2, monitored by mass spectrometry, was found to be dissociated).
H + NO2 → OH + NO
F + H2O → OH + HF
Figure 2.
Configuration of the flow reactor used in the measurements of SH yield in reaction (1).

OH radicals were monitored at m/z = 96 and 98 (HOBr+) after being scavenged with an excess of Br2 ([Br2] = (3–5) × 1013 molecule cm−3, added in the end of the reactor 5 cm upstream of the sampling cone (Figure 1):
k4 = 2.16 × 10−11 exp(207/T) cm3 molecule−1 s−1 (T = 220–950 K) [17]. The reaction of OH with Br2 was also used for the absolute calibration of HOBr signals linking [HOBr] formed to the consumed fraction of [Br2], [OH] = [HOBr] = Δ[Br2]. A similar detection scheme was employed for the SH radicals, the product of reaction (1). SH was transformed to stable species HSBr in reaction with Br2 and monitored by mass spectrometry at m/z = 112 and 114 (HSBr+):
k5 = 5.7 × 10−11 exp(160/T) cm3 molecule−1 s−1 (T = 273–373 K) [18].
OH + Br2 → HOBr + Br
SH + Br2 → HSBr + Br
Carbonyl sulfide was delivered to the reactor from a flask with a known gaseous OCS/He mixture and was detected by mass spectrometry at its parent peak of m/z = 60 (OCS+). The absolute concentrations of OCS as well as of other stable species (Br2, NO2, F2, H2, and H2S) were derived from their flow rates from monometrically prepared mixtures.
The purities of the gases used were as follows: He (>99.9995%, Alphagaz, Air Liquide France Industrie, Paris, France), passed through a liquid nitrogen trap; H2 (>99.998%, Alphagaz); F2, 5% in helium (Alphagaz); Br2 (>99.99%, Aldrich, St. Louis, MO, USA); NO2 (>99%, Alphagaz); H2S > 99.5% (Alphagaz); OCS (15% in He, Messer, Bad Soden, Germany) > 99.99%.
3. Results
3.1. Rate Constant of Reaction (1)
The measurements of k1 were carried out at a total pressure of 2 Torr and in the presence of NO2 ((2–5) × 1013 molecule cm−3) in the reactor in order to minimize the impact of the possible secondary reaction of OH with SH radical, which was identified as a primary reaction product:
OH + SH → products
To our knowledge, the data on the rate constant of reaction (6) are not available in the literature, but it is expected to be rapid. Indeed, we observed an increase in the rate of OH consumption at lower NO2 concentrations (~2 × 1012 molecule cm−3), in line with similar observations of Leu and Smith [5]. In the presence of NO2, SH radicals are scavenged:
k7 = 2.9 × 10−11 exp(240/T) cm3 molecule−1 s−1 (T = 221–415 K) [19]. HSO radicals formed in reaction (7) are in turn trapped by NO2
k8 = 9.6 × 10−12 cm3 molecule−1 s−1 (T = 296 K) [20].
SH + NO2 → HSO + NO
HSO + NO2 → HSO2 + NO
Experiments were performed under pseudo-first-order conditions in a large excess of OCS (0.26 × 1014 < [OCS] < 51.5 × 1014 molecule cm−3) over the initial concentration of OH radicals, [OH]0 = (1–3) × 1011 molecule cm−3. Examples of linear semilogarithmic plots of the OH signal against reaction time are shown in Figure 3, where the line for [OCS] = 0 represents the kinetics of OH loss on the surface of the flow reactor. The pseudo-first-order rate constants, k1′ = k1 × [OCS] + kw (kw is the rate of heterogeneous loss of OH radicals), were derived from the fit of the experimental data to the following equation: ln([OH]0/[OH]) = k1′ × t, where t is the reaction time. Determined in this way, values of k1′ were corrected (usually a few percent correction and up to 24% in a few kinetic runs) for OH diffusion [21].
A linear least-squares fit of the k1′ data as a function of [OCS] at each temperature provides the rate constant of reaction (1). Figure 4 shows examples of such plots at a few temperatures. The Y-intercepts in Figure 4 are in the range (15 ± 5) s−1, in good agreement with the values of kw measured in the absence of OCS in the reactor. All the results obtained for k1 are summarized in Table 1.
One of the difficulties linked to measuring the rate constant of reaction (1), mentioned several times in previous studies, is the presence of H2S impurities in the OCS. H2S reacts much more quickly with OH than OCS, and, therefore, even a very low concentration of H2S can lead to OH consumption comparable to that in the slow reaction of OH with OCS
k9 = 7.95 × 10−20 T2.68exp(783/T) cm3 molecule−1 s−1 (T = 228–518 K) [18]. The purity of the OCS used in the present work was >99.99%, as stated by the supplier. It was not possible to detect such a low impurity of H2S using mass spectrometry since the mass spectrum of OCS contains a relatively high peak at m/z = 34, corresponding to the 34S sulfur isotope. For a possible H2S impurity of 0.01% in OCS and with k9 calculated from the above expression, the contribution of reaction (9) to the values of k1 determined in the present study is estimated to decrease from 7% at T = 365 K to 0.7% at T = 960 K. Ultimately, the possible influence of this side reaction on the current measurements was considered negligible, and no correction for k1 was made.
OH + H2S → H2O + SH
Another possible complication that could impact the OH kinetics is the recycling of OH, especially at the highest temperatures of the study. As noted above, the measurements of k1 were conducted in the presence of NO2 in the reactor, which leads to the formation of HSO2 in the sequence of reactions (7) and (8). This species is unstable at elevated temperatures and dissociates through different pathways depending on its structure [22]:
H atoms formed in reaction (10) could recycle OH radicals in the presence of NO2 in the reactive system. The computed by Goumri et al. [22] low pressure limit data for the rate constant of reaction (10) results in the rates of HSO2 decomposition of (75–800) s−1 in the temperature range (700–960) K and 2 Torr total pressure. Although the reaction of H atoms with OCS can somewhat attenuate the OH recycling process (if it occurs), nevertheless, at the maximum temperatures of the study, the impact on the OH decays in reaction (1) is expected to be significant:
k12 = 6.6 × 10−13 (T/298)3 exp(−1150/T) cm3 molecule−1 s−1 (T = 255–1830 K) [13]. However, it should be noted that in the measurements of k1 at high temperatures, we have not observed any evidence for this OH regeneration: The OH decays were exponential, and the rate of OH consumption did not slow down with the reaction time; at sufficiently high concentrations of OCS, the total consumption of OH was observed. In addition, at two temperatures, T = 545 and 755 K, we have carried out the measurements of k1 in an NO2-free system using reaction (3) as a source of OH radicals ([OH]0 ≤ 1.0 × 1011 molecule cm−3). The measured values of k1 were in good agreement with those obtained in the presence of NO2 (Table 1). This observation also seems to indicate that the possible hypothetical recycling of OH through reactions (10) and (2) is of minor importance, at least under the experimental conditions of the present measurements.
HSO2 + M → H + SO2 + M
HSOO + M → SH + O2 + M
H + OCS → CO + SH
The theoretical studies [8,10] predict a pressure dependence of the overall rate constant of reaction (1). In order to explore the dependence of k1 on pressure, in addition to the bulk of the measurements at a total pressure of 2 Torr, we performed rate constant measurements at P = 1 and 8 Torr at T = 500 and 495 K, respectively. The results of these experiments are presented in Figure 5. The slopes of the straight lines in Figure 5 provide k1 (±2σ) = (2.96 ± 0.09) × 10−14 and (3.04 ± 0.09) × 10−14 cm3 molecule−1 s−1 at P = 1 and 8 Torr, respectively, in good agreement with each other and with 2 Torr data (Table 1), indicating that k1 does not depend on pressure, at least around T = 500 K.
3.2. Products of Reaction (1)
In this series of experiments, we measured the yield of SH radical, which was identified as a primary reaction product. The measurements were carried out using a relative method, which allowed us to avoid the complex procedure of measuring the absolute concentrations of OH and SH radicals. The configuration of the flow reactor used in these experiments is shown in Figure 2. Let us note that in this case, OH radicals were formed in a reaction of F atoms with H2O: an NO2-free system was needed in order to avoid SH loss in reaction (7). The following protocol was employed in the measurements. First, OH radicals ([OH]0 ≈ 3 × 1011 molecule cm−3) supplied through the movable injector reacted with Br2 (reaction 4, [Br2] ≈ 3 × 1012 molecule cm−3) in the main reactor, producing HOBr ([HOBr]0). Then, OCS was introduced in the reactor, and the same concentration of OH was titrated with a mixture of Br2 and OCS, resulting in the formation of HOBr ([HOBr]) and SH in competing reactions (4) and (1), respectively. In the presence of Br2, SH radicals are converted to HSBr according to reaction (5). In the experiments, the concentration of OCS was varied ([OCS] = (0.5 − 20) × 1014 molecule cm−3), and the concentration of the formed HSBr ([HSBr]) was measured as a function of the fraction of OH consumed upon the addition of OCS ([HOBr]0 − [HOBr]). Similar experiments were carried out with the addition of H2S instead of OCS in the reactor. In this way, the yield of SH in reaction (1) could be measured relative to that in reaction of OH radicals with H2S (reaction 9), where the yield of SH is 100%. The measurements were carried out at two temperatures, 500 and 750 K. Results are shown in Figure 6. The slopes of the straight lines (linear through origin fit to the experimental data) in Figure 6, corresponding to the relative yield of SH in reactions of OH with OCS and H2S, are similar to within 2%. These results seem to clearly indicate that the SH + CO2-forming channel of reaction (1) is the major one with a branching ratio close to unity, at least at temperatures between 500 and 750 K.
The relative method applied in these experiments is very convenient, not only because there is no need to measure absolute concentrations of radicals but also because it minimizes the possible impact of side reactions (such as, for example, SH wall loss or SH + SH reaction) on the results of the measurements. The point is that experiments with both compounds (OCS and H2S) are carried out under the same conditions (OH and SH concentrations, reaction time), so any possible relative change in SH concentration due to the side process will be the same in both chemical systems. Additional SH production in secondary reactions seems unlikely. The Br atom formed in reactions (4) and (5) is the only active species present in the reactor. In principle, SH radicals can be formed in the reaction of the Br atom with H2S,
k13 = 1.4 × 10−11exp(−2752/T) cm3 molecule−1 s−1 (T = 319–431 K) [23]. However, considering extrapolated values of k13 = 5.7 × 10−14 and 3.6 × 10−13 cm3 molecule−1 s−1 at T = 500 and 750 K, respectively, and the range of the H2S concentrations used ([H2S]max ≈ 7 × 1012 and 5 × 1012 molecule cm−3 at T = 500 and 750 K, respectively), the potential impact of this process can be neglected.
Br + H2S → SH + HBr
4. Discussion
4.1. Temperature Dependence of k1
All the data available for the rate constant of reaction (1) are shown in Figure 7. In earlier studies, using flash photolysis-resonance fluorescence technique, only upper limits of the reaction rate constants were reported: k1 < 0.7 × 10−14 and <2 × 10−14 cm3 molecule−1 s−1 at T = 299 and 430 K, respectively, by Atkinson et al. [2] and k1 < 0.88 × 10−14, <1.29 × 10−14 and <3.27 × 10−14 cm3 molecule−1 s−1 at T = 298, 343 and 369 K, respectively, by Ravishankara et al. [4]. In contrast, Kurylo [3] measured a much higher value of the rate constant: k1 = (5.66 ± 1.21) × 10−14 cm3 molecule−1 s−1 at T = 296 K. Wahner and Ravishankara [7] minimized complications due to the secondary reactions of photolysis products of OCS that were believed [4] to impact the measurements of Kurylo [3] and determined k1 = (1.92 ± 0.25) × 10−15 cm3 molecule−1 s−1 at T = 298 K. In two studies [5,6] the rate constant of reaction (1) was measured using a discharge flow apparatus combined with the resonance-fluorescence method for OH detection. Leu and Smith [5] reported the Arrhenius expression k1 = 1.3 × 10−12 exp(−(2300 ± 100)/T) cm3 molecule−1 s−1 in the temperature range 300–517 K. Cheng and Lee [6] determined k1 over the temperature range 255–483 K: k1 = 1.13 × 10−13 exp(−(1200 ± 400)/T) cm3 molecule−1 s−1. Finally, in a most recent study, Schmidt et al. [8] applied a relative-rate technique and measured k1 = (5.3 ± 3.6) 10−15 cm3 molecule−1 s−1 at 296 K and 700 Torr total pressure.
Current measurements of k1 were conducted in the temperature range of 365–960 K. At lower temperatures in the flow reactor, it was problematic to accurately measure the rate constant of the slow reaction, especially in the presence of the considerable heterogeneous OH loss (~10 s−1). The fit to the present measurements of k1 (solid black line in Figure 7) yields the following Arrhenius expression:
at T = 365–960 K. We estimate this expression to be accurate within the total relative uncertainty of 20% (including statistical and systematic errors) over the investigated temperature range.
k1 = (2.35 ± 0.25) × 10−12 exp(−(2144 ± 56)/T) cm3 molecule−1 s−1,
The present results can be compared to those of previous studies conducted in a temperature range that overlaps with the temperature range of the present work. Our measurements are consistent with the upper limits reported for k1 by Atkinson et al. [2] and Ravishankara et al. [4], but are 2 to 3 times higher than the k1 data reported in discharge flow/resonance-fluorescence studies [5,6], although the temperature dependence measured for k1 in the present work is similar to that reported by Leu and Smith [5]. Such a significant difference is difficult to explain, especially since all three studies were carried out in similar flow reactors and under similar experimental conditions. The only difference between these works is the OH detection method. Unfortunately, only the original experimental data at T = 517 K are presented in the paper of Leu and Smith [5]. However, the analysis of these data raises some comments. The authors used a flow reactor with a fixed radical source, fixed detector, and moveable injector for OCS. In this configuration, plots of k1′ versus [OCS] should not result in the positive intercept observed by the authors. This observation may be due to some unaccounted side processes. Considering this fact, as well as the rather low values of k1′ in the experiments, the final results for k1 are expected to be highly uncertain. It seems that at low temperatures, the measurements were even more complicated due to the greater scatter of data noted by the authors. A similar comment appears to be valid for the work of Cheng and Lee [6], where very low pseudo-first-order rate constants were measured, for example, k1′ < 15 s−1 at T = 273 and 255 K; ≤31 s−1 at T = 300 K.
It can be noted that the activation energy (4.26 kcal mol−1) measured in the present work is very similar to the energy barrier calculated for the addition of OH to OCS in theoretical studies of reaction (1): 4.4 [8], 4.4 [9], and 4.2 kcal mol−1 [10].
Interestingly, the extrapolation of the present data for k1 to lower temperatures (dashed line in Figure 7) results in k1 = (1.76 ± 0.35) × 10−15 cm3 molecule−1 s−1 at T = 298 K, in excellent agreement with the absolute measurements of Wahner and Ravishankara [7] and Cheng and Lee [6]. The current recommendation for k1 by the NASA Panel for Data Evaluation, k1 = 7.2 × 10−14 exp(−1070/T) cm3 molecule−1 s−1 (T = 255–423 K, red solid line in Figure 7), is based on k1 measured by Wahner and Ravishankara [7] at T = 298 K and the temperature dependence reported by Cheng and Lee [6]. In this work, a significantly stronger dependence of the rate constant on temperature is observed. In terms of atmospheric implications, this does not change anything: the present work confirms that the OH + OCS reaction is very slow and, as a consequence, is of minor importance in atmospheric chemistry.
Theoretical calculations of Saheb et al. [10] (not shown in Figure 7) predict the total rate constants that are lower by more than three orders of magnitude than measured experimentally. In this regard, it should be noted that the rate constants computed by Saheb et al. [10] for the SH-forming channel of reaction (1) are included in a detailed chemical kinetic model for the oxidation of carbonyl sulfide developed by Glarborg and Marshall [11]. It should be kept in mind that, for example, at T = 500 and 960 K, the recommended theoretical values for k1 are lower by a factor of 4 × 105 and 1200, respectively, than reported in the present work.
4.2. Pressure Dependence of k1
The present measurements of k1 at total pressures from 1 to 8 Torr and T = 500 K indicate that the rate constant does not depend on pressure under these conditions. This result supports previous experimental observations. Cheng and Lee [6] reported no observable change in the rate constant at pressures between 0.9 and 5.9 Torr at room temperature. The room temperature measurements of Wahner and Ravishankara [7] also demonstrated the lack of an effect of the total pressure and of the buffer gas on the reaction rate constant. These experimental findings are in clear contradiction with theoretical calculations that predict a significant dependence of k1 on total pressure. Thus, Schmidt et al. [8] calculated that at room temperature, the rate constant of reaction (1) increases by a factor of two with increasing pressure from 10 to 700 Torr. An even more pronounced increase in the rate constant with pressure was suggested in the theoretical work of Saheb et al. [10]. Thus, according to their calculations, the rate constant increases almost linearly with pressure under the conditions of the present experiments (T = 500 K and P = 1–8 Torr), which contradicts the experiment. It can be stated that there is a clear discrepancy between the available theoretical and experimental data on the dependence of k1 on pressure. On the other hand, the question arises as to how appropriate and correct it is to carry out a comparative analysis of experimental and calculated data regarding such a subtle effect as the dependence of the rate constant on pressure when the computed overall rate constants are orders of magnitude lower than the experimental values.
4.3. Products of Reaction (1)
Leu and Smith [5] performed direct mass spectrometric analysis of the reaction products at T = 517 K and concluded that SH is a predominant primary reaction product formed through the mechanism suggested by Kurylo [3]:
where OC(OH)S* is an intermediate excited complex (adduct), which can decompose back to reactants (reaction 14), products (reaction 15), or be collisionally stabilized (reaction 16). They also observed that SH is rapidly oxidized by NO2 to HSO through reaction (7). These findings are in line with the present measurements, resulting in a nearly unity yield of SH in reaction (1) at least at temperatures between 500 and 750 K.
OH + OCS ↔ OC(OH)S*
OC(OH)S* → SH + CO2
OC(OH)S* + M → OC(OH)S + M
Schmidt et al. [8] in their theoretical study suggested two reaction pathways for reaction (1): The pressure-dependent channel forming an energized OC(OH)S*adduct (which can be stabilized or decompose back to reactants) and the pressure-independent CO + SOH-forming channel. The rate constants calculated for the two reaction pathways at T = 300 K are comparable: Independent of pressure k = 3.31 × 10−16 cm3 molecule−1 s−1 for a bimolecular sulfur atom abstraction channel and 0.209 × 10−16, 1.36 × 10−16, and 3.89 × 10−16 cm3 molecule−1 s−1 for the adduct-forming channel at 10, 100, and 700 Torr, respectively. The dissociation of the OC(OH)S* intermediate to CO2 + SH was suggested to be of potential importance only at elevated temperatures. Unfortunately, the range of the elevated temperatures was not specified, which complicates a comparative analysis with the data from this work. Calculations by Schmidt et al. [8] suggest that under atmospheric conditions, the OC(OH)S adduct reacts rapidly with O2 to form CO2 and SOOH, followed by the dissociation of SOOH, resulting in OH recycling. This issue was not explored in this work; the experiments were carried out in the absence of O2 in the reactor ([O2] < 1012 molecule cm−3). In another theoretical study by Saheb et al. [10], reaction (1) has been suggested to proceed through two adduct and SH + CO2-forming channels. The contribution of the SH-forming channel to the overall rate constant was calculated to be nearly 10 and 100% at T = 500 and ≥800 K, respectively. It can be noted that, as in the case of the dependence of the rate constant on temperature and pressure, the theoretical studies do not fit with the experimental data on the reaction products.
Funding
This research was funded by ANR through the PIA (Programme d’Investissement d’Avenir), grant number ANR-10-LABX-100-01.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting reported results are available in this article.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Kettle, A.J.; Kuhn, U.; von Hobe, M.; Kesselmeier, J.; Andreae, M.O. Global budget of atmospheric carbonyl sulfide: Temporal and spatial variations of the dominant sources and sinks. J. Geophys. Res. Atmos. 2002, 107, 4658. [Google Scholar] [CrossRef]
- Atkinson, R.; Perry, R.A.; Pitts, J.N. Rate constants for the reaction of OH radicals with COS, CS2 and CH3SCH3 over the temperature range 299–430 K. Chem. Phys. Lett. 1978, 54, 14–18. [Google Scholar] [CrossRef]
- Kurylo, M.J. Flash photolysis resonance fluorescence investigation of the reactions of OH radicals with OCS and CS2. Chem. Phys. Lett. 1978, 58, 238–242. [Google Scholar] [CrossRef]
- Ravishankara, A.R.; Kreutter, N.M.; Shah, R.C.; Wine, P.H. Rate of reaction of OH with COS. Geophys. Res. Lett. 1980, 7, 861–864. [Google Scholar] [CrossRef]
- Leu, M.-T.; Smith, R.H. Kinetics of the gas-phase reaction between hydroxyl and carbonyl sulfide over the temperature range 300–517 K. J. Phys. Chem. 1981, 85, 2570–2575. [Google Scholar] [CrossRef]
- Cheng, B.-M.; Lee, Y.-P. Rate constant of OH + OCS reaction over the temperature range 255–483 K. Int. J. Chem. Kinet. 1986, 18, 1303–1314. [Google Scholar] [CrossRef]
- Wahner, A.; Ravishankara, A.R. The kinetics of the reaction of OH With COS. J. Geophys. Res. Atmos. 1987, 92, 2189–2194. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Kyte, M.; Østerstrøm, F.F.; Joelsson, L.M.T.; Knap, H.C.; Jørgensen, S.; Nielsen, O.J.; Murakami, T.; Johnson, M.S. On adduct formation and reactivity in the OCS+OH reaction: A combined theoretical and experimental study. Chem. Phys. Lett. 2017, 675, 111–117. [Google Scholar] [CrossRef]
- Danielache, S.O.; Johnson, M.S.; Nanbu, S.; Grage, M.M.L.; McLinden, C.; Yoshida, N. Ab initio study of sulfur isotope fractionation in the reaction of OCS with OH. Chem. Phys. Lett. 2008, 450, 214–220. [Google Scholar] [CrossRef]
- Saheb, V.; Alizadeh, M.; Rezaei, F.; Shahidi, S. Quantum chemical and theoretical kinetics studies on the reaction of carbonyl sulfide with H, OH and O(3P). Comput. Theor. Chem. 2012, 994, 25–33. [Google Scholar] [CrossRef]
- Glarborg, P.; Marshall, P. Oxidation of Reduced Sulfur Species: Carbonyl Sulfide. Int. J. Chem. Kinet. 2013, 45, 429–439. [Google Scholar] [CrossRef]
- Roberts, T.; Dayma, G.; Oppenheimer, C. Reaction rates control high-temperature chemistry of volcanic gases in air. Front. Earth Sci. 2019, 7, 154. [Google Scholar] [CrossRef]
- Bedjanian, Y. Temperature dependent rate constant for the reaction of H-atoms with carbonyl sulfide. Int. J. Chem. Kinet. 2024, 56, 162–167. [Google Scholar] [CrossRef]
- Bedjanian, Y. Experimental Study of the Reaction of O(3P) with Carbonyl Sulfide between 220 and 960 K. J. Phys. Chem. A 2022, 126, 4080–4086. [Google Scholar] [CrossRef] [PubMed]
- Su, M.C.; Kumaran, S.S.; Lim, K.P.; Michael, J.V.; Wagner, A.F.; Harding, L.B.; Fang, D.C. Rate Constants, 1100 ≤ T ≤ 2000 K, for H + NO2 → OH + NO Using Two Shock Tube Techniques: Comparison of Theory to Experiment. J. Phys. Chem. A 2002, 106, 8261–8270. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III—Gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007, 7, 981–1191. [Google Scholar] [CrossRef]
- Bedjanian, Y. Temperature-Dependent Rate Constant for the Reaction of Hydroxyl Radical with 3-Hydroxy-3-methyl-2-butanone. J. Phys. Chem. A 2019, 123, 10446–10453. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, J.B.; Sander, S.P.; Abbatt, J.; Barker, J.R.; Cappa, C.; Crounse, J.D.; Dibble, T.S.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory. Available online: http://jpldataeval.jpl.nasa.gov (accessed on 20 April 2024).
- Wang, N.S.; Lovejoy, E.R.; Howard, C.J. Temperature dependence of the rate constant for the reaction mercapto + nitrogen dioxide. J. Phys. Chem. 1987, 91, 5743–5749. [Google Scholar] [CrossRef]
- Lovejoy, E.R.; Wang, N.S.; Howard, C.J. Kinetic Studies of the Reactions of HSO with NO2, NO, and O2. J. Phys. Chem. 1987, 91, 5749–5755. [Google Scholar] [CrossRef]
- Kaufman, F. Kinetics of elementary radical reactions in the gas phase. J. Phys. Chem. 1984, 88, 4909–4917. [Google Scholar] [CrossRef]
- Goumri, A.; Rocha, J.-D.R.; Laakso, D.; Smith, C.E.; Marshall, P. Characterization of Reaction Pathways on the Potential Energy Surfaces for H + SO2 and HS + O2. J. Phys. Chem. A 1999, 103, 11328–11335. [Google Scholar] [CrossRef]
- Nicovich, J.M.; Kreutter, K.D.; Van Dijk, C.A.; Wine, P.H. Temperature-dependent kinetics studies of the reactions Br(2P3/2) + H2S ↔ SH + HBr and Br(2P3/2) + CH3SH ↔ CH3S + HBr. Heats of formation of SH and CH3S radicals. J. Phys. Chem. 1992, 96, 2518–2528. [Google Scholar] [CrossRef]
Figure 1.
Configuration of the flow reactor used in the measurements of the rate constant of reaction (1).
Figure 1.
Configuration of the flow reactor used in the measurements of the rate constant of reaction (1).

Figure 3.
Example of OH decays in reaction (1) at different concentrations of OCS at T = 520 K and 2 Torr total pressure.
Figure 3.
Example of OH decays in reaction (1) at different concentrations of OCS at T = 520 K and 2 Torr total pressure.

Figure 4.
Plot of pseudo-first-order rate constant, k1′ = k1 × [OCS] + kw, versus concentration of OCS at different temperatures (P = 2 Torr).
Figure 4.
Plot of pseudo-first-order rate constant, k1′ = k1 × [OCS] + kw, versus concentration of OCS at different temperatures (P = 2 Torr).

Figure 5.
Plot of pseudo-first-order rate constant k1′ versus concentration of OCS observed at temperature of nearly 500 K with a total pressure in the reactor of 1 and 8 Torr.
Figure 5.
Plot of pseudo-first-order rate constant k1′ versus concentration of OCS observed at temperature of nearly 500 K with a total pressure in the reactor of 1 and 8 Torr.

Figure 6.
Concentration of SH formed versus consumed concentration of OH ([HOBr]0 − [HOBr], see text) in reactions of OH with OCS and H2S: (a) T = 500 K; (b) T = 750 K.
Figure 6.
Concentration of SH formed versus consumed concentration of OH ([HOBr]0 − [HOBr], see text) in reactions of OH with OCS and H2S: (a) T = 500 K; (b) T = 750 K.
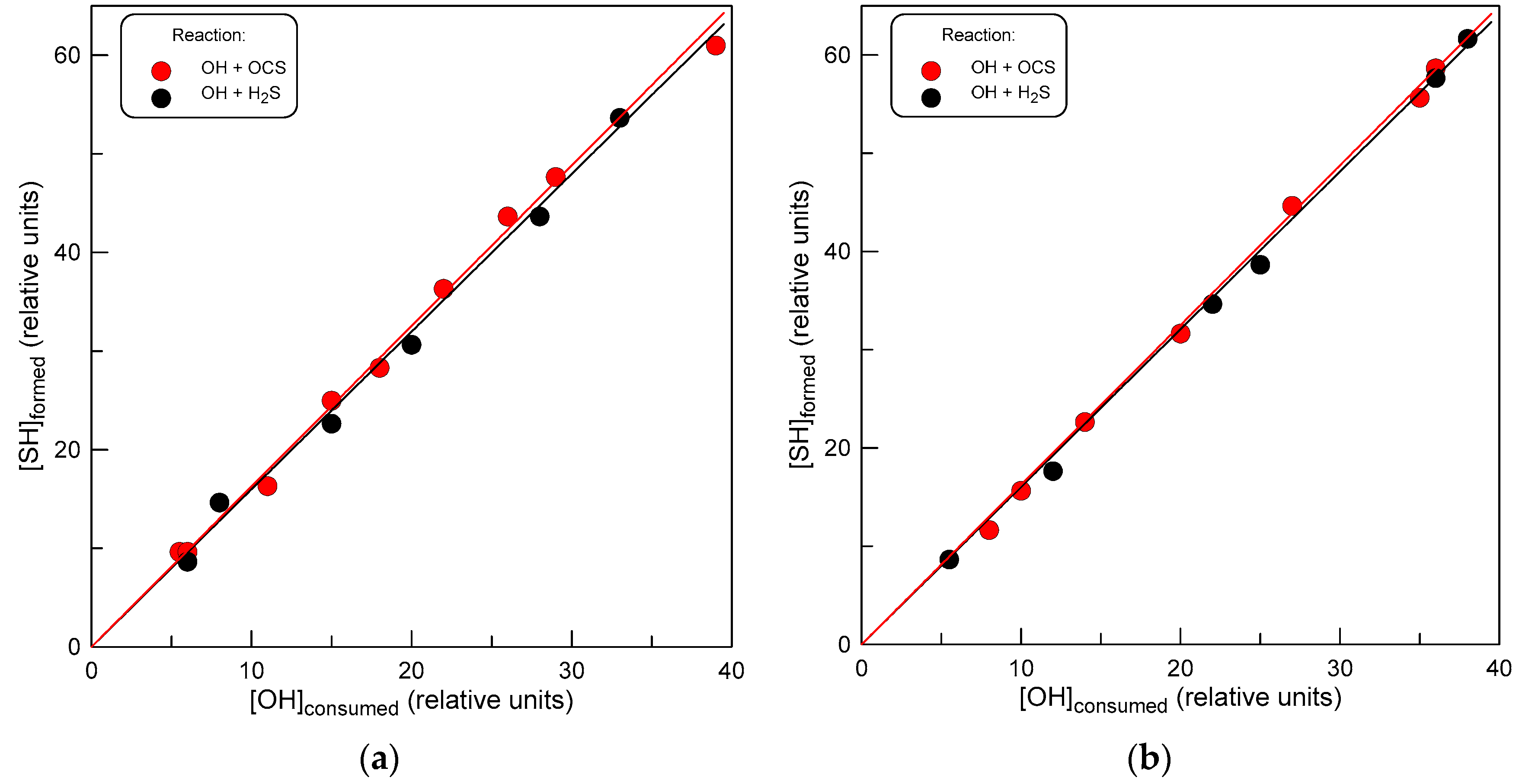
Figure 7.
Temperature dependence of the rate constant of the OH + OCS reaction. Error bars shown for the present measurements of k1 correspond to estimated total uncertainty of 20%. References: Kurylo [3], Atkinson et al. [2], Ravishankara et al. [4], Leu and Smith [5], Cheng and Lee [6], Wahner and Ravishankara [7], Schmidt et al. [8], JPL NASA evaluation [18].
Figure 7.
Temperature dependence of the rate constant of the OH + OCS reaction. Error bars shown for the present measurements of k1 correspond to estimated total uncertainty of 20%. References: Kurylo [3], Atkinson et al. [2], Ravishankara et al. [4], Leu and Smith [5], Cheng and Lee [6], Wahner and Ravishankara [7], Schmidt et al. [8], JPL NASA evaluation [18].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Experimental conditions and results of the present measurements of the rate constant of reaction (1).
Table 1.
Experimental conditions and results of the present measurements of the rate constant of reaction (1).
| T (K) | [OCS] a | k1 b | OH Source | Pressure (Torr) |
|---|---|---|---|---|
| 365 | 3.50–51.5 | 0.69 ± 0.04 | H + NO2 | 2 |
| 385 | 7.19–51.5 | 0.89 ± 0.03 | H + NO2 | 2 |
| 390 | 3.73–50.7 | 0.91 ± 0.02 | H + NO2 | 2 |
| 410 | 2.29–33.6 | 1.24 ± 0.05 | H + NO2 | 2 |
| 440 | 1.45–28.3 | 1.64 ± 0.04 | H + NO2 | 2 |
| 475 | 2.50–31.4 | 2.86 ± 0.09 | H + NO2 | 2 |
| 495 | 3.48–34.1 | 3.04 ± 0.09 | H + NO2 | 8 |
| 500 | 1.96–19.5 | 2.96 ± 0.09 | H + NO2 | 1 |
| 520 | 2.44–33.9 | 4.19 ± 0.08 | H + NO2 | 2 |
| 545 | 2.00–27.7 | 4.62 ± 0.26 | F + H2O | 2 |
| 600 | 1.29–16.1 | 7.27 ± 0.14 | H + NO2 | 2 |
| 675 | 1.16–17.2 | 9.73 ± 0.30 | H + NO2 | 2 |
| 700 | 0.55–14.0 | 10.8 ± 0.34 | H + NO2 | 2 |
| 755 | 3.58–18.1 | 13.7 ± 0.65 | F + H2O | 2 |
| 800 | 0.356–7.12 | 16.4 ± 0.44 | H + NO2 | 2 |
| 880 | 1.42–15.6 | 20.5 ± 0.45 | H + NO2 | 2 |
| 960 | 0.26–9.79 | 23.7 ± 0.34 | H + NO2 | 2 |
a Units of 1014 molecule cm−3. b units of 10−14 cm3 molecule−1 s−1; statistical 2σ uncertainty is given, total estimated uncertainty is 20%.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bedjanian, Y. Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products. Atmosphere 2024, 15, 576. https://doi.org/10.3390/atmos15050576
AMA Style
Bedjanian Y. Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products. Atmosphere. 2024; 15(5):576. https://doi.org/10.3390/atmos15050576
Chicago/Turabian StyleBedjanian, Yuri. 2024. "Experimental Study of the Reaction of OH Radicals with Carbonyl Sulfide between 365 and 960 K: Kinetics and Products" Atmosphere 15, no. 5: 576. https://doi.org/10.3390/atmos15050576
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.