
Retrospective Genotyping of Enteroviruses Using a Diagnostic Nanopore Sequencing Workflow
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Nucleic Acid Extraction
2.3. Enterovirus qPCR
2.4. Enterovirus Amplicon Sequencing
2.5. Bioinformatic Analysis
2.6. Untargeted Whole-Genome Sequencing
2.7. Estimation of Genotype Diversity
3. Results
3.1. Sample Characteristics
3.2. Targeted Amplicon Sequencing and Genotyping
3.3. Untargeted Whole-Genome Sequencing
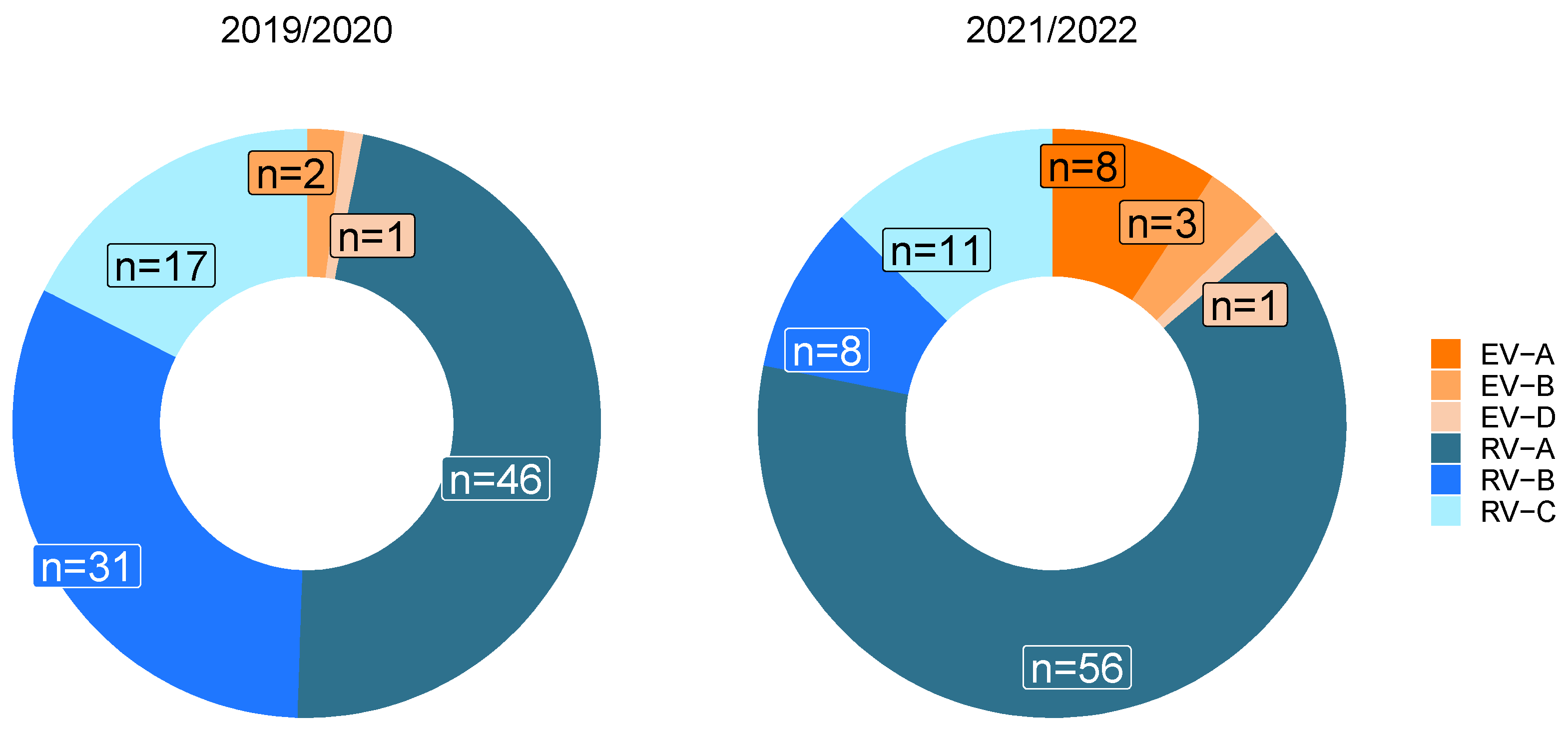
3.4. Influence of the SARS-CoV-2 Pandemic on Circulating Enterovirus Species
3.5. Estimation of Genotype Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hara, M.; Takao, S.; Shimazu, Y. Three-year study of viral etiology and features of febrile respiratory tract infections in Japanese pediatric outpatients. Pediatr. Infect. Dis. J. 2014, 33, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Rihkanen, H.; Ronkko, E.; Nieminen, T.; Komsi, K.L.; Raty, R.; Saxen, H.; Ziegler, T.; Roivainen, M.; Soderlund-Venermo, M.; Beng, A.L.; et al. Respiratory viruses in laryngeal croup of young children. J. Pediatr. 2008, 152, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Nikonov, O.S.; Chernykh, E.S.; Garber, M.B. Enteroviruses: Classification, Diseases They Cause, and Approaches to Development of Antiviral Drugs. Biochemistry 2017, 82, 1615–1631. [Google Scholar] [CrossRef]
- Bubba, L.; Martinelli, M.; Pellegrinelli, L.; Primache, V.; Tanzi, E.; Pariani, E. A 4-year Study on Epidemiologic and Molecular Characteristics of Human Parechoviruses and Enteroviruses Circulating in Children Younger Than 5 Years in Northern Italy. Pediatr. Infect. Dis. J. 2017, 36, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Stellrecht, K.A.; Harding, I.; Woron, A.M.; Lepow, M.L. The impact of an enteroviral RT-PCR assay on the diagnosis of aseptic meningitis and patient management. J. Clin. Virol. 2002, 25 (Suppl. S1), S19–S26. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Moreni, G.; Wolthers, K.C. World-Wide Prevalence and Genotype Distribution of Enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Tan le, V.; Tuyen, N.T.; Thanh, T.T.; Ngan, T.T.; Van, H.M.; Sabanathan, S.; Van, T.T.; Thanh le, T.M.; Nguyet, L.A.; Geoghegan, J.L.; et al. A generic assay for whole-genome amplification and deep sequencing of enterovirus A71. J. Virol. Methods 2015, 215–216, 30–36. [Google Scholar] [CrossRef]
- Dyda, A.; Stelzer-Braid, S.; Adam, D.; Chughtai, A.A. The association between acute flaccid myelitis (AFM) and Enterovirus D68 (EV-D68)—What is the evidence for causation? Euro Surveill. 2018, 23, 17-00310. [Google Scholar] [CrossRef] [PubMed]
- Messacar, K.; Asturias, E.J.; Hixon, A.M.; Van Leer-Buter, C.; Niesters, H.G.M.; Tyler, K.L.; Abzug, M.J. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect. Dis. 2018, 18, e239–e247. [Google Scholar] [CrossRef] [PubMed]
- Janes, V.A.; Minnaar, R.; Koen, G.; van Eijk, H.; Haan, K.D.-D.; Pajkrt, D.; Wolthers, K.C. Presence of human non-polio enterovirus and parechovirus genotypes in an Amsterdam hospital in 2007 to 2011 compared to national and international published surveillance data: A comprehensive review. Euro Surveill. 2014, 19, 20964. [Google Scholar] [CrossRef] [PubMed]
- Bigi, S.; Ramette, A.; Barbani, M.T.; Bieri, A.; Hoffmann, A. Acute flaccid myelitis in Switzerland—Association with enterovirus D68. Swiss Med. Wkly 2023, 153, 40045. [Google Scholar] [CrossRef] [PubMed]
- Bubba, L.; Broberg, E.K.; Jasir, A.; Simmonds, P.; Harvala, H.; Enterovirus study collaborators. Circulation of non-polio enteroviruses in 24 EU and EEA countries between 2015 and 2017: A retrospective surveillance study. Lancet Infect Dis. 2020, 20, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Helfferich, J.; de Lange, M.M.A.; Benschop, K.S.M.; Jacobs, B.C.; Van Leer-Buter, C.C.; Meijer, A.; Bakker, D.P.; de Bie, E.; Braakman, H.M.H.; Brandsma, R.; et al. Epidemiology of acute flaccid myelitis in children in the Netherlands, 2014 to 2019. Euro Surveill. 2022, 27, 2200157. [Google Scholar] [CrossRef] [PubMed]
- Knoester, M.; Helfferich, J.; Poelman, R.; Van Leer-Buter, C.; Brouwer, O.; Niesters, H.G.M.; on behalf of the 2016 EV-D68 AFM Working Group. Twenty-nine cases of Enterovirus-D68–associated acute flaccid myelitis in Europe 2016: A case series and epidemiologic overview. Pediatr. Infect Dis. J. 2019, 38, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.K.; Simmonds, P. The importance of enterovirus surveillance in a post-polio world. Lancet Infect. Dis. 2022, 22, e35–e40. [Google Scholar] [CrossRef] [PubMed]
- Graedel, C.; Ireddy, N.R.; Koch, M.C.; Baumann, C.; Miani, M.A.T.; Barbani, M.T.; Steinlin-Schopfer, J.; Suter-Riniker, F.; Leib, S.L. Genome Sequences of Rare Human Enterovirus Genotypes Recovered from Clinical Respiratory Samples in Bern, Switzerland. Microbiol. Resour. Announc. 2022, 11, e0027622. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.Y.; Hwang, S.; Kim, H.; Song, J.; Ahn, J.; Kang, B.; Kim, K.; Choi, W.; Chung, J.K.; Kim, C.H.; et al. Accuracy of diagnostic methods and surveillance sensitivity for human enterovirus, South Korea, 1999–2011. Emerg. Infect Dis. 2013, 19, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.J.; Lee, J.H.; Kim, M.S.; Bae, G.R.; Jung, C.; Lee, K.; Choi, S.M.; Kim, D.K.; Lee, D.S.; Kim, W.D.; et al. Molecular characterization of enteroviruses detected in Gyeong-Ju and Po-Hang provinces of Korea in 2003. Arch Virol. 2010, 155, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Shukla, D.; Srivastava, S.; Idris, M.Z. High frequency of enterovirus serotype circulation in a densely populated area of India. J. Infect. Dev. Ctries. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lizasoain, A.; Mir, D.; Masachessi, G.; Farias, A.; Rodriguez-Osorio, N.; Victoria, M.; Nates, S. Environmental Surveillance through Next-Generation Sequencing to Unveil the Diversity of Human Enteroviruses beyond the Reported Clinical Cases. Viruses 2021, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Celma, C.; Pegg, E.; Polra, K.; Dunning, J. Detection and Typing of Human Enteroviruses from Clinical Samples by Entire-Capsid Next Generation Sequencing. Viruses 2021, 13, 641. [Google Scholar] [CrossRef] [PubMed]
- Montmayeur, A.M.; Ng, T.F.; Schmidt, A.; Zhao, K.; Magana, L.; Iber, J.; Castro, C.J.; Chen, Q.; Henderson, E.; Ramos, E.; et al. High-Throughput Next-Generation Sequencing of Polioviruses. J. Clin. Microbiol. 2017, 55, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Figas, A. Enteroviruses Associated with Aseptic Meningitis in Poland, 2011–2014. Pol. J. Microbiol. 2016, 65, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, D.R.; Yang, C.F.; Ching, K.; Vincent, A.; Iber, J.; Campagnoli, R.; Mandelbaum, M.; De, L.; Yang, S.J.; Nix, A. Rapid group-, serotype-, and vaccine strain-specific identification of poliovirus isolates by real-time reverse transcription-PCR using degenerate primers and probes containing deoxyinosine residues. J. Clin. Microbiol. 2009, 47, 1939–1941. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Holloway, B.; Dare, R.K.; Kuypers, J.; Yagi, S.; Williams, J.V.; Hall, C.B. Real- time reverse transcription-PCR assay for comprehensive detection of human rhinoviruses. J. Clin. Microbiol. 2018, 46, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Nix, W.A.; Oberste, M.S. Sensitive, semi-nested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- WHO; Centers for Disease Control and Prevention & World Health Organization; Regional Office for Europe. Enterovirus Surveillance Guidelines: Guidelines for Enterovirus Surveillance in Support of the Polio Eradication Initiative; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.D.; Kufner, V.; Schmutz, S.; Boni, J.; Metzner, K.J.; Trkola, A. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 2017, 5, 94. [Google Scholar]
- Kufner, V.; Plate, A.; Schmutz, S.; Braun, D.L.; Günthard, H.F.; Capaul, R.; Zbinden, A.; Mueller, N.J.; Trkola, A.; Huber, M. Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes 2019, 10, 661. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.V.D.; Penaranda, S.; Oberste, M.S.; Vinje, J. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Axmacher, J.C. Estimating the number of species shared by incompletely sampled communities. Ecography 2021, 44, 1098–1108. [Google Scholar] [CrossRef]
- Zou, Y.; Zhao, P. Estimating total species richness: Fitting rarefaction by asymptotic approximation. Ecosphere 2023, 14, e4363. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M.L.; Johnson, K.A. The original Michaelis constant: Translation of the 1913 Michaelis-Menten paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [PubMed]
- Simmonds, P.; Gorbalenya, A.E.; Harvala, H.; Hovi, T.; Knowles, N.J.; Lindberg, A.M.; Oberste, M.S.; Palmenberg, A.C.; Reuter, G.; Skern, T.; et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020, 165, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, O.; Conlan, S.; Deming, C.; Lee-Lin, S.Q.; Huang, X.; NISC Comparative sequencing program; Su, H.C.; Freeman, A.F.; Segre, J.A. Expanded skin virome in DOCK-8-deficient patients. Nat. Med. 2018, 24, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.E.; Black, S. Emerging infectious diseases: A proactive approach. Proc. Natl. Acad. Sci. USA 2017, 114, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.M.; Balalla, S.; Theadom, A.; Jackman, G. A systematic review of the worldwide prevalence of survivors of poliomyelitis reported in 31 studies. BMJ Open 2017, 7, e015470. [Google Scholar] [CrossRef] [PubMed]
- Kufner, V.; Frey, A.C.; Burkhard, S.H.; Schmutz, S.; Ziltener, G.; Zaheri, M.; Wiedmer, C.V.; Plate, A.; Trkola, A.; Huber, M. Exploring viral etiology in upper respiratory tract infections: Insights from mNGS in Swiss outpatients before and during the SARS-CoV-2 pandemic. Swiss Med. Weekly 2024, in press. [Google Scholar]
- Neves, A.; Walther, D.; Martin-Campos, T.; Barbie, V.; Bertelli, C.; Blanc, D.; Bouchet, G.; Erard, F.; Greub, G.; Hirsch, H.H.; et al. The Swiss Pathogen Surveillance Platform—Towards a nation-wide One Health data exchange platform for bacterial, viral and fungal genomics and associated metadata. Microb. Genom. 2023, 9, 001001. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genotype * | 2019/2020 | 2021/2022 | Total |
|---|---|---|---|---|
| Enterovirus A | CVA4 | 0 | 1 | 1 |
| CVA6 | 3 | 10 | 13 | |
| CVA10 | 1 | 6 | 7 | |
| EV-A71 | 2 | 0 | 2 | |
| Enterovirus B | CVA9 | 0 | 2 | 2 |
| CVB1 | 0 | 1 | 1 | |
| CVB2 | 1 | 2 | 3 | |
| CVB3 | 1 | 7 | 8 | |
| CVB4 | 1 | 3 | 4 | |
| CVB5 | 5 | 3 | 8 | |
| E6 | 1 | 2 | 3 | |
| E7 | 3 | 0 | 3 | |
| E11 | 0 | 1 | 1 | |
| E18 | 0 | 2 | 2 | |
| E20 | 2 | 0 | 2 | |
| E21 | 1 | 0 | 1 | |
| E25 | 1 | 1 | 2 | |
| E30 | 1 | 0 | 1 | |
| Enterovirus D | EV-D68 | 1 | 2 | 3 |
| Rhinovirus A | RV-A1 | 6 | 0 | 6 |
| RV-A2 | 1 | 1 | 2 | |
| RV-A8 | 1 | 0 | 1 | |
| RV-A9 | 1 | 0 | 1 | |
| RV-A11 | 3 | 1 | 4 | |
| RV-A12 | 2 | 1 | 3 | |
| RV-A15 | 1 | 3 | 4 | |
| RV-A20 | 2 | 2 | 4 | |
| RV-A21 | 1 | 0 | 1 | |
| RV-A22 | 1 | 0 | 1 | |
| RV-A24 | 1 | 8 | 9 | |
| RV-A25 | 1 | 3 | 4 | |
| RV-A28 | 1 | 0 | 1 | |
| RV-A29 | 0 | 1 | 1 | |
| RV-A30 | 0 | 1 | 1 | |
| RV-A31 | 1 | 4 | 5 | |
| RV-A32 | 1 | 0 | 1 | |
| RV-A33 | 1 | 0 | 1 | |
| RV-A34 | 4 | 1 | 5 | |
| RV-A36 | 1 | 3 | 4 | |
| RV-A39 | 1 | 0 | 1 | |
| RV-A46 | 1 | 3 | 4 | |
| RV-A47 | 2 | 7 | 9 | |
| RV-A49 | 1 | 0 | 1 | |
| RV-A53 | 1 | 3 | 4 | |
| RV-A55 | 1 | 0 | 1 | |
| RV-A56 | 1 | 0 | 1 | |
| RV-A57 | 1 | 1 | 2 | |
| RV-A58 | 0 | 3 | 3 | |
| RV-A59 | 0 | 3 | 3 | |
| RV-A60 | 1 | 0 | 1 | |
| RV-A61 | 0 | 2 | 2 | |
| RV-A65 | 0 | 1 | 1 | |
| RV-A66 | 1 | 0 | 1 | |
| RV-A67 | 1 | 0 | 1 | |
| RV-A71 | 1 | 0 | 1 | |
| RV-A78 | 1 | 2 | 3 | |
| RV-A80 | 1 | 1 | 2 | |
| RV-A82 | 1 | 0 | 1 | |
| RV-A85 | 0 | 2 | 2 | |
| RV-A100 | 1 | 0 | 1 | |
| Rhinovirus B | RV-B3 | 7 | 1 | 8 |
| RV-B4 | 1 | 0 | 1 | |
| RV-B6 | 2 | 0 | 2 | |
| RV-B14 | 5 | 0 | 5 | |
| RV-B26 | 1 | 0 | 1 | |
| RV-B27 | 2 | 2 | 4 | |
| RV-B35 | 1 | 0 | 1 | |
| RV-B42 | 0 | 1 | 1 | |
| RV-B48 | 1 | 0 | 1 | |
| RV-B69 | 1 | 2 | 3 | |
| RV-B70 | 3 | 0 | 3 | |
| RV-B72 | 1 | 1 | 2 | |
| RV-B83 | 0 | 2 | 2 | |
| RV-B84 | 1 | 0 | 1 | |
| RV-B86 | 2 | 0 | 2 | |
| RV-B91 | 2 | 0 | 2 | |
| NA | 1 | 0 | 1 | |
| Rhinovirus C | RV-C1 | 1 | 4 | 5 |
| RV-C5 | 0 | 1 | 1 | |
| RV-C7 | 1 | 0 | 1 | |
| RV-C11 | 1 | 2 | 3 | |
| RV-C12 | 1 | 0 | 1 | |
| RV-C15 | 2 | 0 | 2 | |
| RV-C17 | 1 | 0 | 1 | |
| RV-C24 | 1 | 0 | 0 | |
| RV-C25 | 0 | 1 | 1 | |
| RV-C26 | 1 | 0 | 1 | |
| RV-C33 | 1 | 0 | 1 | |
| RV-C41 | 2 | 0 | 2 | |
| RV-C42 | 1 | 1 | 2 | |
| RV-C43 | 2 | 0 | 2 | |
| RV-C45 | 1 | 1 | 2 | |
| RV-C51 | 0 | 1 | 1 | |
| RV-C53 | 1 | 0 | 1 | |
| RV-C56 | 0 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Ackeren, V.; Schmutz, S.; Pichler, I.; Ziltener, G.; Zaheri, M.; Kufner, V.; Huber, M. Retrospective Genotyping of Enteroviruses Using a Diagnostic Nanopore Sequencing Workflow. Pathogens 2024, 13, 390. https://doi.org/10.3390/pathogens13050390
van Ackeren V, Schmutz S, Pichler I, Ziltener G, Zaheri M, Kufner V, Huber M. Retrospective Genotyping of Enteroviruses Using a Diagnostic Nanopore Sequencing Workflow. Pathogens. 2024; 13(5):390. https://doi.org/10.3390/pathogens13050390
Chicago/Turabian Stylevan Ackeren, Vanessa, Stefan Schmutz, Ian Pichler, Gabriela Ziltener, Maryam Zaheri, Verena Kufner, and Michael Huber. 2024. "Retrospective Genotyping of Enteroviruses Using a Diagnostic Nanopore Sequencing Workflow" Pathogens 13, no. 5: 390. https://doi.org/10.3390/pathogens13050390