
Copper Resistance Mechanism and Copper Response Genes in Corynebacterium crenatum

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Cultivation Conditions, and Plasmids
2.2. RNA Preparation and Transcriptome Sequencing
2.3. Differentially Expressed Gene (DEG) Analysis and Functional Annotation
2.4. DNA Manipulation and Strain Construction
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Cell Growth Tests
2.7. Copper Adsorption
2.8. ATP and NADP/NADPH Analysis
2.9. Bioinformatics and Statistical Analysis
3. Results
3.1. DEGs and Enrichment Analysis
3.2. Effects of Copper on Energy Metabolism
3.3. Effects of Copper on Metal Ion Transport
3.4. Cell-Surface Display of Copper-Binding Peptides Influencing Growth Rate in Copper Stress
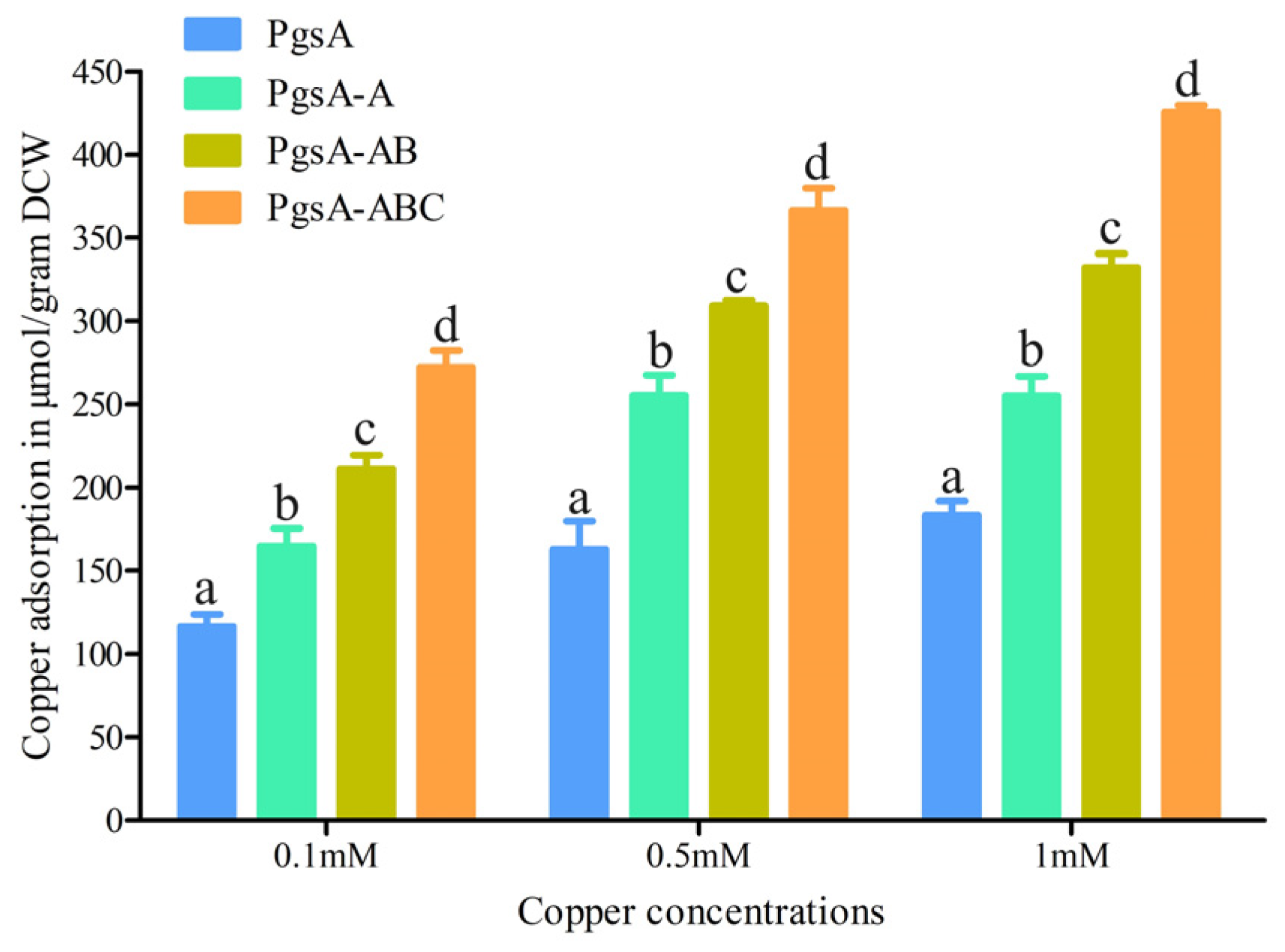
3.5. Copper Adsorption
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, P.H.; Lin, C.; Guo, K.H.; Yeh, Y.C. Development of a pigment-based whole-cell biosensor for the analysis of environmental copper. RSC Adv. 2017, 7, 29302. [Google Scholar] [CrossRef]
- Blake, D.; Nar, M.; D’Souza, N.A.; Glenn, J.B.; Klaine, S.J.; Roberts, A.P. Treatment with coated layer double hydroxide clays decreases the toxicity of copper-contaminated water. Arch. Environ. Contam. Toxicol. 2014, 66, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gu, J.D. A unified conceptual framework involving maintenance energy, metabolism and toxicity involved in research on degradation of environmental organic pollutants. Int. Biodeterior. Biodegrad. 2021, 162, 105253. [Google Scholar] [CrossRef]
- Gu, J.D. Bioremediation of toxic metals and metalloids for cleaning up from soils and sediments. Appl. Environ. Biotechnol. 2018, 3, 48–51. [Google Scholar]
- Bontidean, I.; Lloyd, J.R.; Hobman, J.L.; Wilson, J.R.; Csoregi, E.; Mattiasson, B.; Brown, N.L. Bacterial metal-resistance proteins and their use in biosensors for the detection of bioavailable heavy metals. J. Inorg. Biochem. 2000, 79, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Mao, J.; Hu, M.; Wang, X.Y.; Huo, M.X. Responses to copper stress in the metal-resistant bacterium Cupriavidus gilardii CR3: A whole-transcriptome analysis. J. Basic Microbiol. 2019, 59, 446–457. [Google Scholar] [CrossRef]
- Kershaw, C.J.; Brown, N.L.; Constantinidou, C.; Patel, M.D.; Hobman, J.L. The expression profile of Escherichia coli K-12 in response to minimal, optimal and excess copper concentrations. Microbiology 2005, 151, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Quintana, J.; Novoa-Aponte, L.; Argüello, J.M. Copper homeostasis networks in the bacterium Pseudomonas aeruginosa. J. Biol. Chem. 2017, 292, 15691–15704. [Google Scholar] [CrossRef]
- Pant, G.; Garlapati, D.; Agrawal, U.; Prasuna, R.G.; Mathimani, T.; Pugazhendhi, A. Biological approaches practised using genetically engineered microbes for a sustainable environment: A review. J. Hazard. Mater. 2021, 5, 124631. [Google Scholar] [CrossRef]
- Saravanan, A.; Kumar, P.S.; Ramesh, B.; Srinivasan, S. Removal of toxic heavy metals using genetically engineered microbes: Molecular tools, risk assessment and management strategies. Chemosphere 2022, 298, 134341. [Google Scholar] [CrossRef]
- Huang, M.; Zhao, Y.; Feng, L.; Zhu, L.F.; Zhan, L.; Chen, X.L. Role of ClpB from Corynebacterium crenatum in thermal stress and arginine fermentation. Front. Microbiol. 2020, 11, 1660. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhao, Y.; Feng, L.; Zhu, L.F.; Zhan, L.; Chen, X.L. Role of the ClpX from Corynebacterium crenatum involved in stress responses and energy metabolism. Appl. Microbiol. Biotechnol. 2020, 104, 5505–5517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ren, L.; Yu, M.; Zhou, Y.; Ye, B. Enhanced L-ornithine production by systematic manipulation of l-ornithine metabolism in engineered Corynebacterium glutamicum S9114. Bioresour. Technol. 2018, 250, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mashruwala, A.A.; Eilers, B.J.; Fuchs, A.L.; Norambuena, J.; Earle, C.A.; Guchte, A.; Tripet, B.P.; Copié, V.; Boyd, J.M. The ClpCP complex modulates respiratory metabolism in Staphylococcus aureus and is regulated in a SrrAB-dependent manner. J. Bacteriol. 2019, 201, e00188-19. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Wang, Y.; Selvamani, V.; Yoo, I.; Kim, T.W.; Hong, S.H. A novel strategy for the microbial removal of heavy metals: Cell-surface display of peptides. Biotechnol. Bioprocess Eng. 2021, 26, 1–9. [Google Scholar] [CrossRef]
- Cai, R.; Jiang, Y.; Yang, W.; Yang, W.; Shi, S.; Shi, C.; Hu, J.; Gu, W.; Ye, L.; Zhou, F.; et al. Surface-displayed IL-10 by recombinant Lactobacillus plantarum reduces Th1 responses of RAW264.7 cells stimulated with Poly(I:C) or LPS. J. Microbiol. Biotechnol. 2016, 26, 421–431. [Google Scholar] [CrossRef]
- Gordon, A.S.; Howell, L.D.; Harwood, V. Responses of diverse heterotrophic bacteria to elevated copper concentrations. Can. J. Microbiol. 1994, 40, 408–411. [Google Scholar] [CrossRef]
- Boyd, S.M.; Rhinehardt, K.L.; Ewunkem, A.J.; Harrison, S.H.; Thomas, M.D.; Graves, J.L., Jr. Experimental evolution of copper resistance in Escherichia coli produces evolutionary trade-offs in the antibiotics chloramphenicol, bacitracin, and sulfonamide. Antibiotics 2022, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Stephanie, S.; Daniela, Z.; Boris, L.; Michael, B.; Melanie, B.; Ben, A. The two-component signal transduction system CopRS of Corynebacterium glutamicum is required for adaptation to copper-excess stress. PLoS ONE 2011, 6, e22143. [Google Scholar]
- Osman, O.; Tanguichi, H.; Ikeda, K.; Park, P.; Tanabe-Hosoi, S.; Nagata, S. Copper-resistant halophilic bacterium isolated from the polluted Maruit Lake, Egypt. J. Appl. Microbiol. 2010, 108, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Wiesemann, N.; Bütof, L.; Herzberg, M.; Hause, G.; Berthold, L.; Etschmann, B.; Brugger, J.; Martinez-Criado, G.; Dobritzsch, D.; Baginsky, S.; et al. Synergistic toxicity of copper and gold compounds in Cupriavidus metallidurans. Appl. Environ. Microbiol. 2017, 83, e01679-17. [Google Scholar] [CrossRef]
- Cidre, I.; Pulido, R.P.; Burgos, M.J.G.; Gálvez, A.; Lucas, R. Copper and zinc tolerance in bacteria isolated from fresh produce. J. Food Prot. 2017, 80, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, L.; Dussan, J. Biosorption and bioaccumulation of heavy metals on dead and living biomass of Bacillus sphaericus. J. Hazard. Mater. 2009, 167, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Khemiri, I.; Tebbji, F.; Sellam, A. Transcriptome analysis uncovers a link between copper metabolism, and both fungal fitness and antifungal sensitivity in the opportunistic yeast Candida albicans. Front. Microbiol. 2020, 11, 935. [Google Scholar] [CrossRef]
- Gong, H.; Gao, Y.; Zhou, X.; Xiao, Y.; Wang, W.W.; Tang, Y.T.; Zhou, S.; Zhang, Y.Y.; Ji, W.X.; Yu, L. Cryo-EM structure of trimeric Mycobacterium smegmatis succinate dehydrogenase with a membrane-anchor SdhF. Nat. Commun. 2020, 11, 4245. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Liu, W.; Qin, C.; Xu, Y.; Zhou, X.; Wen, Q.; Ma, W.; Huang, Y.; Chen, X. Copper Resistance Mechanism and Copper Response Genes in Corynebacterium crenatum. Microorganisms 2024, 12, 951. https://doi.org/10.3390/microorganisms12050951
Huang M, Liu W, Qin C, Xu Y, Zhou X, Wen Q, Ma W, Huang Y, Chen X. Copper Resistance Mechanism and Copper Response Genes in Corynebacterium crenatum. Microorganisms. 2024; 12(5):951. https://doi.org/10.3390/microorganisms12050951
Chicago/Turabian StyleHuang, Mingzhu, Wenxin Liu, Chunyan Qin, Yang Xu, Xu Zhou, Qunwei Wen, Wenbin Ma, Yanzi Huang, and Xuelan Chen. 2024. "Copper Resistance Mechanism and Copper Response Genes in Corynebacterium crenatum" Microorganisms 12, no. 5: 951. https://doi.org/10.3390/microorganisms12050951