


Genes 2024, 15(5), 593; https://doi.org/10.3390/genes15050593 - 07 May 2024
Abstract
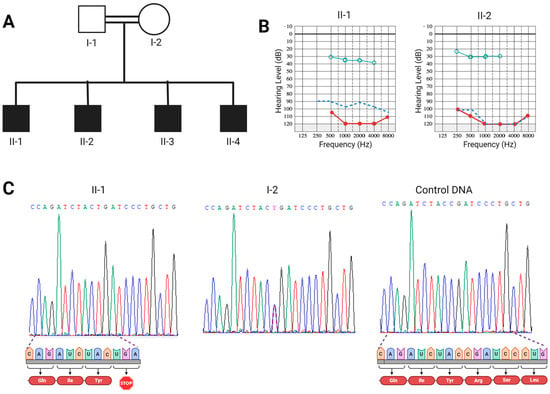
Background: Alport syndrome (AS) is a common and heterogeneous genetic kidney disease, that often leads to end-stage kidney disease (ESKD). Methods: This is a single-center, retrospective study that included 36 adults with type IV collagen (COL4) mutations. Our main scope was to describe
[...] Read more.
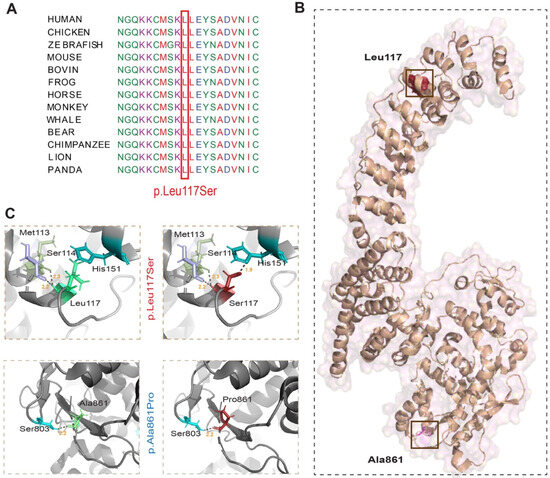
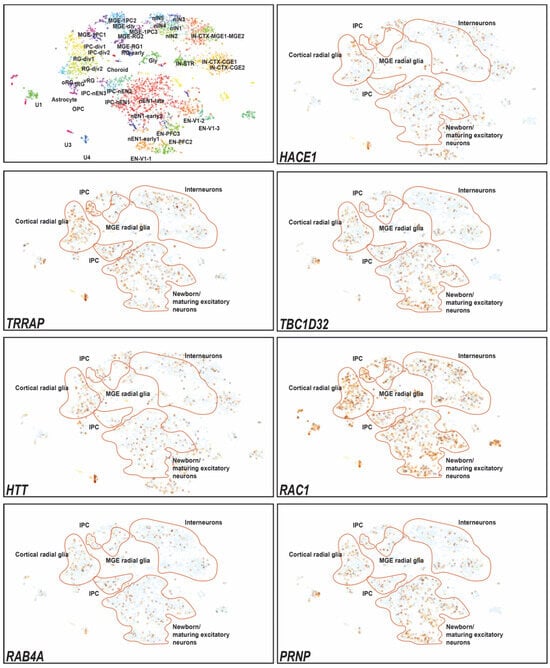
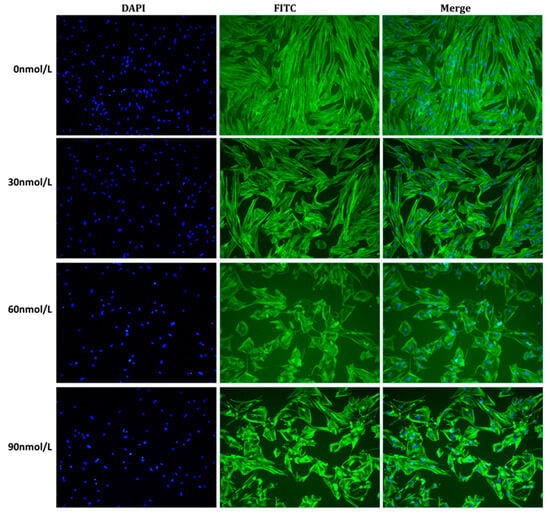
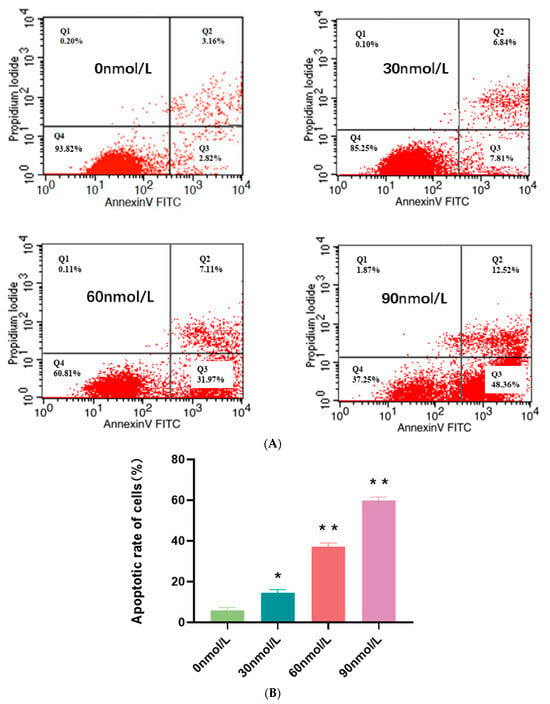
Background: Alport syndrome (AS) is a common and heterogeneous genetic kidney disease, that often leads to end-stage kidney disease (ESKD). Methods: This is a single-center, retrospective study that included 36 adults with type IV collagen (COL4) mutations. Our main scope was to describe how genetic features influence renal survival. Results: A total of 24 different mutations were identified, of which eight had not been previously described. Mutations affecting each of the type IV collagen α chains were equally prevalent (33.3%). Most of the patients had pathogenic variants (61.1%). Most patients had a family history of kidney disease (71%). The most prevalent clinical picture was nephritic syndrome (64%). One-third of the subjects had extrarenal manifestations, 41.6% of patients had ESKD at referral, and another 8.3% developed ESKD during follow-up. The median renal survival was 42 years (95% CI, 29.98–54.01). The COL4A4 group displayed better renal survival than the COL4A3 group (p = 0.027). Patients with missense variants had higher renal survival (p = 0.023). Hearing loss was associated with lower renal survival (p < 0.001). Conclusions: Patients with COL4A4 variants and those with missense mutations had significantly better renal survival, whereas those with COL4A3 variants and those with hearing loss had worse prognoses.
Full article
(This article belongs to the Special Issue Genetic and Phenotypic Correlation: Gene–Disease Validation Series II)









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}