
meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives
by
 and
and
Daniel Aicher
1,
Dinusha Damunupola
2,
Christian B. W. Stark
1,*,†,
Arno Wiehe
1,*,‡ and
Christian Brückner
2,* 1
Institut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany
2
Department of Chemistry, University of Connecticut, 55 N Eagleville Rd., Storrs, CT 06269-3060, USA
*
Authors to whom correspondence should be addressed.
†
Current address: Institute of Organic Chemistry, Department of Chemistry, Universität Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany.
‡
Current address: Biolitec Research GmbH, Otto-Schott-Str. 15, 07745 Jena, Germany.
Molecules 2024, 29(9), 2144; https://doi.org/10.3390/molecules29092144
Submission received: 12 April 2024
/
Revised: 30 April 2024
/
Accepted: 1 May 2024
/
Published: 5 May 2024
(This article belongs to the Special Issue Porphyrin-Based Compounds: Synthesis and Application, 2nd Edition)
Abstract
:meso-Tetrahexylporphyrin was converted to its corresponding 7,8-dihydroxychlorin using an osmium tetroxide-mediated dihydroxylation strategy. Its diol moiety was shown to be able to undergo a number of subsequent oxidation reactions to form a chlorin dione and porpholactone, the first meso-alkylporphyrin-based porphyrinoid containing a non-pyrrolic building block. Further, the diol chlorin was shown to be susceptible to dehydration, forming the porphyrin enol that is in equilibrium with its keto-chlorin form. The meso-hexylchlorin dione could be reduced and it underwent mono- and bis-methylation reactions using methyl-Grignard reagents, and trifluoromethylation using the Ruppert-Prakash reagent. The optical and spectroscopic properties of the products are discussed and contrasted to their corresponding meso-aryl derivatives (where known). This contribution establishes meso-tetrahexyl-7,8-dihydroxychlorins as a new and versatile class of chlorins that is susceptible to a broad range of conversions to generate functionalized chlorins and a pyrrole-modified chlorin analogue.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Hydroporphyrins–chlorins (dihydroporphyrins) and bacteriochlorins (tetrahydroporphyrins) are nature’s principle light harvesting pigments [1]. Synthetic hydroporphyrins and hydroporphyrin analogues have been prepared due to their fundamental importance in the understanding of these photosynthetic pigments [2], for use in bioimaging [3], as markers in fluorescence-guided surgery [4,5,6], as photosensitizers in the photodynamic [3,7] or photothermal [8] therapy of tumors, as photoantimicrobials [9,10], as light-harvesting dyes in dye-sensitized solar cells [11], as catalysts [12], and for many other biological and technical applications [13,14,15,16,17]. The vast majority of synthetic porphyrins (and hydroporphyrins) fall into two classes: the β-alkylporphyrins, modeled closely after nature’s β-alkyltetrapyrroles, and the popular meso-arylporphyrins that have no direct precedent in nature.
Hydroporphyrins can be prepared by the conversion of porphyrins, by total synthesis, or by the modification of tetrapyrroles derived from nature [13,14,15,16,17]. One well-established method to convert porphyrins to chlorins is through their OsO4-mediated dihydroxylation. This versatile method is suitable for converting β-alkylporphyrins 1 or meso-arylporphyrins 3 to their corresponding 7,8-dihydroxychlorins 2 and 4, respectively (Scheme 1) [18,19,20,21,22,23,24]. The regiochemistry and other mechanistic aspects of this reaction are well understood [25,26]. Importantly, the diol functionality in the dihydroxychlorins can be used as a synthetic handle for a number of subsequent functional group transformations, generating a host of chlorins and chlorin analogues [19,20,24,27,28,29,30].
Curiously, the chemistry of meso-alkylporphyrins (5) (Figure 1), although known for decades [31,32,33], has been far less studied, by a wide margin, compared to that of their β-alkyl- or meso-aryl analogues [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50]. meso-Alkyl-porphyrins have also not been, outside of the patent literature [51,52], converted to chlorins. In fact, reports on meso-alkylchlorins, in general, are rare: the lone example is meso-tetramethylchlorin and its metal complexes, such as meso-tetramethylchlorin nickel complex 6 [32,53,54]. The meso-tetramethylchlorin metal complexes were formed as by-products during the synthesis of its corresponding metalloporphyrin by means of a 4 × 1-type metal-templated condensation of a suitably derivatized pyrrole [32,53,54].
We recently reported on a neutral tetraalkylporphyrin with high solubility in aqueous solutions [50]. This porphyrin is potentially attractive for biomedical applications. An increase in the intensity of its absorption spectrum in the red region of its optical spectrum and/or a bathochromic shift would much increase its applicability. Typically, the desired optical properties shift is achieved by the conversion of a porphyrin to a chlorin [16,55]. The osmium tetroxide-mediated dihydroxylation of a porphyrin is irreversible, produces a chlorin, and, by virtue of the introduction of the diol functionality, can be expected to increase the amphiphilicity of the molecule, a benefit for its application in photomedicine [43,56,57]. However, neither this dihydroxylation reaction nor the synthetic manipulation of the diol moiety have been, outside of the patent literature [51,52], applied to any meso-alkylporphyrins.
This contribution reports the OsO4-mediated dihydroxylation and subsequent diol functional group manipulations of an archetype meso-tetraalkylporphyrin, meso-tetrahexylporphyrin. This work identifies meso-alkyl-7,8-dihydroxychlorins as a new class of chlorins that are susceptible to a broad range of conversions to generate functionalized chlorins and a pyrrole-modified chlorin analogue.
2. Results and Discussion
2.1. The Osmylation of meso-Tetrahexylporphyrin 7
The OsO4-mediated dihydroxylation of meso-tetrahexylporphyrin 7 took place under standard conditions that are also applicable to octaalkylporphyrins and meso-tetraphenylporphyrin: 1–2 equiv OsO4 in CHCl3/pyridine (optionally applied in two aliquots) over several days at ambient temperature, followed by the reductive cleavage of the initially formed osmate ester (Scheme 2). We found that the osmylation takes place at a comparable, or possibly slightly lower rate, as in meso-tetraphenylporphyrin, a finding in line with our expectations for the electron-rich porphyrin 7 [26].
The success of the dihydroxylation reaction is indicated by the formation of the main polar product 8 with the characteristic optical properties of a chlorin, coupled with a double Soret band (Figure 2). Product 8 can be isolated in satisfactory yields. Its 1H NMR spectrum shows a loss of the four-fold symmetry of the starting porphyrin and the formation of a two-fold symmetric product, with diagnostic pyrroline proton signals (at 6.1 ppm); elemental analysis and ESI+ HR mass spectrometry confirmed its expected composition (reproductions of key spectra of all new compounds are provided as Supplementary Material).
Parallel to the dihydroxylation of meso-tetraarylporphyrins [58,59], an ‘over-oxidized’ tetrahydroxybacteriochlorin derivative 9 was also observed as a minor side product. A single isomer of bacteriochlorin 9 was spectroscopically characterized, but the relative stereochemistry of its two cis-diol functionalities was not assigned.
2.2. The Transformations of meso-Tetrahexyl-7,8-dihydroxychlorin 8
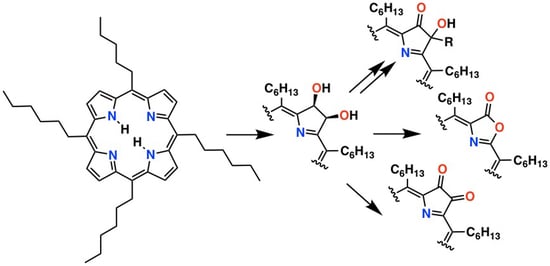
We, and others, have demonstrated the versatility of the diol functionality of octaalkyl- and meso-tetraphenylchlorin diols with respect to their functional group transformation, generating a number of porphyrin and chlorin analogues [19,20,27,28,29]. We were now also able to demonstrate the applicability of some of these reactions to meso-tetrahexyl-7,8-dihydroxychlorin 8 (Scheme 3).
The oxidation of the meso-tetraarylchlorin diols to their corresponding diones using a range of oxidants is well known [28,60]. Using Dess–Martin periodinane (DMP), this transformation is also applicable to chlorin diol 8, forming meso-tetrahexylchlorin dione 10. The success of the reaction was demonstrated by the loss of the pyrroline hydrogen signals in the 1H NMR spectrum of product 10, its characteristically broadened optical spectrum, and its composition (as per ESI+ HR-MS). The formation of side products and the overall relatively low yield of the dione could be rationalized by the formation of side products resulting from the oxidation of the α-position of its meso-alkyl chains (see below). An alternate path to dione 10 was more successful (the conversion of 11 to 10, see Scheme 3 and below).
The (adventitious) acid-catalyzed or thermally driven dehydration of meso-tetraarylchlorin diols [61,62], as well as the related pinacol–pinacolone rearrangement of octaalkychlorin diols [19,27,63], have been previously described. Accordingly, the treatment of chlorin diol 8 with acid induces this dehydration reaction in a satisfying yield, generating ketone-chlorin 11. An inspection of the 1H NMR spectrum of free base 11 (CHCl3, 25 °C) shows that the equilibrium position of this keto-enol tautomerism lies, at a minimum, in the ratio of 10:1 on the side of the ketone-chlorin 11A over its tautomeric form, enol-porphyrin 11B. This differs from the position of equilibrium for its corresponding meso-tetraphenyl derivative; metalation or solvents play a large role in the establishment of this equilibrium [64,65]. Irrespective of the presence of a ketone form in the tautomeric mixture, 11 is inert to reactions with Grignard or alkyl lithium reagents. It is, however, highly susceptible to oxidation with the DMP, smoothly providing dione 10.
The reduction of ketone/enol 11 with NaBH4 forms mono-β-hydroxychlorin 12, characterized by its chlorin-like optical spectrum (Figure 3) and presence of a complex set of peaks assigned to three non-equivalent pyrroline hydrogen atoms (that are, in part, also coupled to the -OH proton). Its corresponding meso-tetraphenylchlorin was observed to form as a side product during the hydrogen sulfide reduction of the diol osmate ester [58], or by an alumina-catalyzed (oxygen-mediated) oxidation of its corresponding tetrahydrochlorin [66].
Over the years, we have developed the cetyltrimethylammonium permanganate (CTAP)-induced oxidation of meso-tetraarylporphyrins or meso-tetraaryldihydroxychlorins to form their corresponding porpholactones [67,68,69,70]. This conversion is complementary to a number of other oxidation reactions that form porpholactones [71,72,73,74,75]. When a meso-hexylchlorin diol was reacted with CTAP under standard conditions, the polar diol converted to form non-polar compound 13 with a porphyrin-like optical spectrum (Figure 2). Its 1H NMR spectrum indicates the presence of six non-equivalent β-pyrrole protons, and its diagnostic composition (as per ESI+ HRMS) highlights the loss of a carbon atom (and the uptake of two oxygen atoms). Both its FTIR and {1H}13C NMR spectra show signatures for the presence of a carbonyl functionality (νC=O at 1842 cm−1 and a peak at 168 ppm, respectively). All data support the formation of the target meso-hexylporpholactone 13, the first example of a meso-alkylporphyrin analogue containing a non-pyrrolic heterocycle [16,76,77,78].
2.3. Direct Oxidations of meso-Tetrahexylporphyrin
In the reactions of diol chlorin 8 or keto-enol chlorin 11 with DMP (Scheme 3), we noticed the formation of several side products with porphyrin-like optical spectra. A slight variation of the reaction solvent used, when applied to meso-tetrahexylporphyrin 7, provided the main product 14 in good yield (Scheme 4). Its composition indicated that a single CH2-to-C=O oxidation had taken place. Its 1H NMR indicated the presence of all pyrrole hydrogens, but the region corresponding to the most shielded CH2 group, the group closest to the ring, had become more complex, suggestive of the formation of three non-equivalent meso-hexyl groups, supporting its assignment as the 1’-oxohexyl derivative 14. Evidently, the porphyrin ring activated the meso-hexyl group to allow for an alkane CH oxidation, a reaction not ordinarily observed in DMP-mediated oxidations [79,80].
A number of direct—i.e., not requiring any prior porphyrin functionalization—porphyrin-to-porpholactone conversions are known; chief among them are the RuCl3/oxone®/bipy oxidation developed by Zhang and co-workers [75,81] and the CTAP reaction referred to above [69,70,82]. Both reactions can be applied to meso-tetrakis(pentafluorophenyl)porphyrins, but the CTAP oxidation is ineffective for the oxidation of meso-tetraphenylporphyrins [70]. We were thus surprised to find that both RuCl3/oxone®/bipy oxidation and CTAP-mediated oxidation are applicable to the oxidation of free base meso-tetrahexylporphyrin 7 to generate the target meso-tetrahexylporpholactone 13. In fact, CTAP-mediated oxidation is highly efficient and rapid (5 min), generating porpholactone 13 in a high yield and with only a modicum of side products, even outperforming Ru-based oxidation (or a two-step oxidation via chlorin diol 8, Scheme 3).
The direct (and clean) porphyrin-to-porpholactone conversion achieved using CTAP is also possible for the previously reported 5,15-dihexylporphyrin 15 [41,45] (Scheme 5). Here, two compounds of near-identical UV–vis spectra and identical compositions (as per ESI+ HRMS), but with very differing quantities, are formed. The major compound 16A could be fully spectroscopically characterized and its 1H NMR spectrum, for example, supports its assignment as the target dihexylporpholactone, but the second compound 16B was only formed in insufficient quantities to fully characterize it by NMR spectroscopy. Nonetheless, based on the identical UV–vis and mass spectra of the two compounds, we assigned them to be the two possible lactone regioisomers 16A and 16B.
The assignment of these specific regioisomers can be accomplished using the heteronuclear three-bond correlation HMBC (3JC,H) spectrum of the major fraction, isomer 16A (Figure 4): In the {1H}13C NMR spectrum of 16A, the most down-field-shifted quaternary carbon signal at 168 ppm can be assigned to the carbonyl carbon atom; likewise, the two most down-field-shifted signals in its 1H NMR spectrum (s at 9.94 and 9.84 ppm) stand out and can be clearly assigned to the two non-equivalent meso-hydrogen atoms. A clear three-bond interaction between the carbonyl carbon of the lactone moiety and one of the meso-protons (at 9.94 ppm) can be seen in the HMBC spectrum of 16A, unequivocally identifying this porpholactone as the regioisomer 16A, shown carrying its carbonyl group on the side of the (less sterically encumbered) meso-position; the other isomer 16B would not be expected to show any 3JC,H coupling between the carbonyl carbon atom and any meso-hydrogen atom. The sterically less encumbered orientation of the lactone moiety was also the prevalent orientation found in 5,15-diphenylporpholactones [68].
2.4. The Transformations of meso-Tetrahexylchlorin-7,8-dione 10
The carbonyl-type reactivity of the ketone groups in meso-tetraarylchlorin-7,8-diones has been amply demonstrated [30,71,83,84]. We were able to confirm this also for meso-alkyldione 10. Correspondingly, dione 10 could be reduced with NaBH4 to its corresponding diol 17 possessing a UV–vis spectrum that is near-identical to that of diol 9. Both compounds possess the same composition (as per HR-MS). We suggest that diol 17 is the trans-diol isomer of the cis-isomer 9 (Scheme 6). The 1H NMR spectrum of the 2-fold rotationally symmetric trans-isomer 17 and its mirror-symmetric cis-isomer 9 vary slightly, most significantly with respect to a 0.5 ppm difference in the shift of the pyrroline proton; their Rf-values differ also, with diol 17 being less polar than diol 9.
Dione 10 is also susceptible to single and double methyl-Grignard addition, forming α-hydroxyketone 18 in satisfying yields and (trans) diol 19 in moderate yields, even under more forcing conditions. This reaction has precedent in the meso-arylporphyrin series [85]. α-Hydroxyketone 18 can be reduced with NaBH4 to the corresponding (likely trans) diol 20. Dione 10 also undergoes nucleophilic CF3-group addition using the Ruppert-Prakash reagent, CF3SiMe/TBAF [86,87], yielding product 18F, without showing the formation of a bis-adduct. Like α-hydroxyketone 18, the ketone moiety in its trifluoromethyl analogue 18F can also be reduced to form the corresponding chlorin diol 20F. All novel compounds had the expected analytical and spectroscopic properties. All diols (17, 19, 20, and 20F) show very similar chlorin-like UV–vis spectra (see, e.g., Figure 5 and ESI); the strong electronic influence of the β-ketone is clearly visible. These compounds add to the series of hydroporphyrins with a redox state between that of the dione and the diol chlorin, some of which were explored in the meso-aryl series as well [88].
In summary, using meso-tetrahexylporphyrin 7 as a representative of the much under-studied meso-alkylporphyrins, we have shown that it is readily converted to a range of chlorins and porpholactone using multiple reactions, most of which had precedents in the chemistry of the better-investigated meso-aryl- and β-alkylporphyrins. The key steps are the smooth OsO4-mediated dihydroxylation of the porphyrin to its corresponding dihydroxychlorin, which functionalizes the porphyrin toward further transformations. Other direct oxidative conversions of the porphyrin and of the meso-hexyl group could also be demonstrated, with some reaction features unique to the meso-alkylporphyrin/chlorin. Thus, this contribution establishes meso-tetrahexyl-7,8-dihydroxychlorins as a new and versatile class of chlorins that is susceptible to a broad range of conversions to generate functionalized chlorins and pyrrole-modified chlorin analogues. The meso-alkylporphyrin and -chlorins are readily derived, soluble, and stable, ideal prerequisites for further work on this class of compounds.
3. Materials and Methods
3.1. Materials
Aluminum-backed, silica gel 60, 250 μm thick analytical plates were used for analytical TLC; either 20 × 20 cm, glass-backed, silica gel 60, 500 µm thick preparative TLC plates or standard grade, 60 Å, 32–63 µm flash column silica gel were used for the chromatographic separation and purification of the products.
3.2. Instruments
1H and {1H}13C NMR spectra were recorded on Bruker instruments in the solvents indicated and were referenced to residual solvent peaks or internal TMS. Where present, structural assignments were performed with the help of COSY (3JH,H), HMQC (1JC,H), and HMBC (2JC,H and 3 JC,H) spectra and NOE experiments. UV–vis spectra were recorded either on Cary 50, 60, or 100 (Varian, Palo Alto, CA, USA, now Agilent, Santa Clara, CA, USA) or Specord S300 UV-vis (Analytik Jena, Jena, Germany) spectrometers in 1 cm glass or quartz cells, in the solvents indicated. Fluorescence emission spectra were recorded on a Cary Eclipse spectrometer in 1 cm glass or quartz cells, in the solvent indicated. FT-IR spectra were recorded on an Alpha (Bruker, Billerica, MA, USA) instrument (diamond ATR). Mass spectrometry analyses were performed on a QStar Elite (AB Sciex, Framingham, MA, USA) Quadrupole-TOF, Agilent 6210 ESI-TOF (Agilent, Santa Clara, CA, USA), or Ionspec QFT-7 ESI-FTICR (Varian Inc., Lake Forest, CA, USA) mass spectrometer.
3.3. General Procedures
3.3.1. General Procedure A: Hydride Reduction
The starting porphyrin is dissolved in CH2Cl2/MeOH and, at 0 °C, 5–10 equivalents of NaBH4 are added (in portions). Stirring continued until the TLC control indicated the consumption of the starting material. Water was added to the reaction mixture and the organic phase was separated in a separatory funnel. If the aqueous phase was colored, it was extracted with CH2Cl2 or ethyl acetate. The combined organic phases were washed with water, dried over Na2SO4 (anhyd), and the solvent removed by rotary evaporation. The residue was chromatographed and the fractions recrystallized.
3.3.2. General Procedure B: CTAP Oxidation
The starting porphyrin or chlorin was, in a round-bottom flask equipped with a stir bar, dissolved in CH2Cl2, and freshly prepared CTAP was added and stirred at ambient conditions. The reaction’s progress was observed by TLC; once the desired conversion had taken place, the solvent was reduced by rotary evaporation, the concentrate passed through a pad of Celite®, and the pad was washed with CH2Cl2 until the eluent was largely colorless. The combined filtrates were evaporated to dryness by rotary evaporation and the crude mixture was chromatographed and the fractions recrystallized.
3.3.3. General Procedure C: DMP Oxidation
The starting porphyrin was dissolved in CH2Cl2 and a solution of 5–6 equivalents of Dess–Martin periodinane (DMP) dissolved in CH2Cl2 was added drop-wise in ambient conditions until the TLC control indicated the consumption of the starting material. Water was added to the reaction mixture and the organic phase was separated in a separatory funnel. If the aqueous phase was colored, it was extracted with CH2Cl2 or ethyl acetate. The combined organic phases were washed with water, dried over Na2SO4 (anhyd), and the solvent removed by rotary evaporation. The residue was chromatographed and the fractions recrystallized.
3.4. Osmium Tetroxide-Mediated Dihydroxylation of meso-Tetrahexylporphyrin (7): Formation of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8) and meso-Tetrahexyl-7,8,7,18-cis-tetrahydroxybacteriochlorin (9)
In a 50 mL round-bottom flask, meso-tetrahexylporphyrin (7) (148 mg, 0.2 mmol, 1 equiv.) was dissolved in a mixture of CHCl3 (15 mL, EtOH-stabilized) and freshly distilled pyridine (2 mL). A solution of OsO4 (2.9 mL of an OsO4 stock solution of 1.0 g OsO4, 3.93 mmol, in 25 mL of 30% pyridine/CHCl3, amounting to 0.46 mmol, 2.3 equiv.) was added to the mixture. [CAUTION: note the hazard and risk of using OsO4; the use of a fume hood and suitable PPE—nitrile gloves, safety goggles, and a lab coat—are required.] The flask was stoppered, shielded from light with aluminum foil, and magnetically stirred at ambient temperature for ~7 days. The progress of the reaction was monitored by occasional TLC for the consumption of the starting material. Once no further progress was noted, the solvent was removed to dryness on a rotary evaporator at the lowest temperature feasible. The crude osmate ester product was then dissolved in a solution of 10% MeOH/CHCl3 (~15 mL) and vigorously stirred with a sat. aqueous (or 1:1 MeOH/H2O) NaHSO3 solution (~20 mL) for up to 7 days (monitored by TLC). Once all the intermediate osmate ester was consumed, the mixture was extracted with CHCl3 twice, and its organic fraction was isolated and dried over Na2SO4 anhyd. The drying agent was removed by filtration and the filtrate was evaporated to dryness by rotary evaporation. The resulting residue was dissolved in a minimal amount of CH2Cl2 and loaded onto a silica gel column and eluted with CH2Cl2. The first fraction, eluted with 90% hexanes/CH2Cl2, was starting material 7 (~5%). Target chlorin diol 8 was eluted with 30% hexanes/CH2Cl2. Alternatively, silica gel column chromatography using 95% CH2Cl2/5% ethyl acetate is suitable. Slow evaporation from a hexanes/CH2Cl2 mixture (or recrystallization from CH2Cl2:MeOH) provided product 8 as a purple fluffy solid (101 mg, 75%). A second, more polar light pink fraction was identified as tetrahydroxybacteriochlorin 9.
8: Rf (silica–20% hexanes/CH2Cl2) = 0.69; Rf (silica–95% CH2Cl2/5% ethyl acetate) = 0.85. 1H NMR (400 MHz; CDCl3): δ −1.87–2.11 (br s, 1H), 0.92–0.89 (m, 6H), 1.55–1.37 (m, 8H), 1.70 (ddq, J = 36.0, 14.5, 7.2 Hz, 4H), 2.10–1.99 (m, 2H), 2.38 (dq, J = 14.4, 7.4 Hz, 2H), 2.76–2.58 (m, 1H), 4.11 (t, J = 8.0 Hz, 2H), 4.56–4.40 (m, 2H), 6.11 (s, 1H), 8.73 (d, J = 4.9 Hz, 1H), 8.95 (d, J = 4.8 Hz, 1H), 9.12 (s, 1H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ 159.7, 152.0, 139.5, 133.9 (α-C), 129.6 (C-17, C-18), 124.7, 121.4 (C-2, C-3, C-12, C-13), 110.8 (meso-C), 73.0 (C-7, C-8), 38.1 (C-32), 36.2 (C-26), 35.0 (C-31), 32.9 (C-25), 31.9, 31.9 (C-28, C-34), 30.4, 30.2 (C-27, C-34), 22.9, 22.9 (C-29, C-35), 14.3, 14.3 (C-30, C-36) ppm. UV–vis (CH2Cl2) λmax (log ε): 402 (5.31), 423 (sh), 522 (3.07), 550 (3.12), 588 (3.45), 640 (2.39) nm. Fl (λexcitation = λSoret) λmax (rel. intensity) = 652 (1.0), 720 (0.09) nm. MS (EI, 170 °C): m/z = 680 (39%, [M]+), 662 (100%, [M − H2O]+), 646 (21%, [M–2 OH]+), 609 (21%, [M–C5H11]+), 591 (45%, [M − H2O–C5H11]+). HR-MS (ESI+, 100% CH3CN, 30 V cone voltage, TOF detection): m/z calc’d for C44H65N4O2 [M + H]+, 681.5108; found 681.5134.
9: 1H NMR (500 MHz, DMSO-d6): δ = –1.83 (s, 2H, NH), 0.92 (t, J = 7.3 Hz, 12 H, 4 × CH3), 1.33–1.40 (m, 8 H, 4 × CH2), 1.42–1.48 (m, 8 H, 4 × CH2), 1.68 (m, 8 H, 4 × CH2), 2.04–2.18 (m, 8 H, 4 × CH2), 4.15–4.21 (m, 4 H, 2 × CH2), 4.35–4.42 (m, 4 H, 2 × CH2), 5.72–5.76 (m, 4 H, -OH), 6.22–6.25 (m, 4 H, pyrroline-H), 8.92–8.94 (m, 4 H, pyrrole-H) ppm. {1H}13C NMR (126 MHz, DMSO-d6): δ = 14.05 (CH3), 22.27 (CH2), 29.69 (CH2), 30.69 (CH2), 31.38 (CH2), 32.61 (CH2), 35.52 (CH2), 72.56 (pyrroline-C), 113.42 (meso-C), 120.47 (β-C), 134.97 (α-C), 158.82 (α-C) ppm. UV–vis (CH2Cl2): λmax (log ε): 373 (5.26), 413 (4.26), 507 (3.92), 539 (4.68), 650 (3.77), 704 (4.77) nm. HRMS (ESI, TOF detection): m/z calc’d. for C44H67N4O4+ ([M + H]+): 715.5157; found 715.5153.
3.5. DMP Oxidation of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8) or meso-Tetrahexylchlorin-7-one (11): Formation of meso-Tetrahexylporphyrin-7,8-dione (10)
According to General Procedure C, meso-tetrahexyl-7,8-cis-dihydroxychlorin (8) or meso-tetrahexylchlorin-7-one (11) (250 mg, 0.37 mmol), in CH2Cl2 (15 mL), was reacted with a 15% (w/w) solution of Dess–Martin periodinane (DMP) in CH2Cl2 (1.6 g, 1.9 mmol) over 3 h in ambient conditions. Column chromatography on silica–2:1 CH2Cl2/hexane. Recrystallization from CH2Cl2/MeOH provided the dione 10 as a purple solid in 53% yield (133 mg).
10: 1H NMR (500 MHz, CDCl3): δ –2.60 (s, 2 H, NH), 0.94–0.97 (m, 12 H, 4 × CH3), 1.36–1.53 (m, 16 H, 8 × CH2), 1.69–1.80 (m, 8 H, 4 × CH2), 1.95–2.03 (m, 4 H, 2 × CH2), 2.34–2.41 (m, 4 H, 2 × CH2), 4.35–4.41 (m, 4 H, 2 × CH2), 4.58–4.63 (m, 4 H, 2 × CH2), 9.00–9.02 (m, 2 H, β-H), 9.11–9.13 (m, 4 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.30 (CH3), 14.34 (CH3), 22.89 (CH2), 22.94 (CH2), 30.22 (CH2), 30.41 (CH2), 31.15 (CH2), 31.97 (CH2), 32.01 (CH2), 35.56 (CH2), 36.44 (CH2), 38.35 (CH2), 114.35 (meso-C), 122.80 (meso-C), 124.76 (β-C), 125.28 (β-C), 131.66 (β-C), 136.78 (α-C), 138.47 (α-C), 139.48 (α-C), 154.41 (α-C), 189.22 (β-CO) ppm. UV–vis (CH2Cl2): λmax (log ε): 406 (5.31), 491 (4.23), 715 (3.72) nm. HRMS (ESI+, TOF detection): m/z calc’d for C44H61N4O2+ ([M + H]+): 677.4789; found: 677.4793. HRMS (ESI−, TOF detection): m/z calc’d for C44H59N4O2− ([M − H]−): 675.4644; found: 675.4621.
3.6. Dehydration of meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8): Formation of meso-Tetrahexylchlorin-7-one (11)
Dihydroxychlorin 8 (400 mg, 0.59 mmol) was dissolved in TFA (30 mL) and heated to 65 °C [CAUTION: Trifluoroacetic acid is a strongly corrosive acid that poses an inhalation hazard. The use of a fume hood and suitable PPE—nitrile gloves, safety goggles, and a lab coat—are required]. After 8 h, the mixture was added to ice water in a separatory funnel and a 20% aqueous solution of NaOH was added until neutrality was reached. The product is extracted with ethyl acetate (2×), the combined organic phases were washed with water, dried over Na2SO4 (anhyd.), and the solvent removed by rotary evaporation. Recrystallization of the residue from CH2Cl2/MeOH delivered the final product 11 as a purple solid in 96% yield (375 mg).
11: 1H NMR (500 MHz, CDCl3): (keto tautomer, 10:1 dominant over the enol tautomer) δ −2.49 (s, 1 H, NH), −2.26 (s, 1 H, NH), 0.94–0.99 (m, 12 H, 4 × CH3), 1.35–1.55 (m, 16 H, 8 × CH2), 1.61–1.67 (m, 2 H, CH2), 1.73–1.83 (m, 6 H, 3 ×CH2), 2.00–2.14 (m, 4 H, 2 × CH2), 2.37–2.48 (m, 4 H, 2 × CH2), 3.77–3.81 (m, 2 H, CH2), 4.51 (s, 2 H, β-H), 4.62–4.72 (m, 6 H, 3 × CH2), 8.84 (d, J = 4.8 Hz, 1 H, β-H), 9.13 (d, J = 4.7 Hz, 1 H, β-H), 9.16 (d, J = 4.8 Hz, 1 H, β-H), 9.19 (d, J = 4.6 Hz, 1 H, β-H), 9.22 (d, J = 4.5 Hz, 1 H, β-H), 9.25 (d, J = 4.6 Hz, 1 H, β-H) ppm. {1H}13C NMR (126 MHz, DMSO-d6): δ =14.25 (CH3), 14.27 (CH3), 14.33 (CH3), 22.86 (CH2), 22.96 (CH2), 30.17 (CH2), 30.34 (CH2), 30.44 (CH2), 30.81 (CH2), 31.88 (CH2), 32.00 (CH2), 32.06 (CH2), 35.04 (CH2), 35.18 (CH2), 35.72 (CH2), 35.78 (CH2), 36.09 (CH2), 38.22 (CH2), 38.39 (CH2), 46.40 (β-C), 109.56 (meso-C), 116.00 (meso-C), 119.46 (meso-C), 122.86 (β-C), 123.12 (meso-C), 123.65 (β-C), 124.41 (β-C), 125.64 (β-C), 130.26 (β-C), 131.34 (β-C), 135.30 (α-C), 136.41 (α-C), 138.36 (α-C), 138.45 (α-C), 145.96 (α-C), 152.36 (α-C), 154.57 (α-C), 155.13 (α-C), 205.62 (β-CO) ppm. UV–vis (CH2Cl2): λmax (log ε): 419 (5.39), 433 (5.29), 534 (4.20), 570 (4.29), 604 (4.00), 659 (3.76) nm. HRMS (ESI+, TOF detection): m/z calc’d for C44H63N4O+ ([M + H]+): 663.4996; found: 663.4937.
3.7. Hydride Reduction of meso-Tetrahexylchlorin-7-one (11): Formation of meso-Tetrahexyl-7-hydroxychlorin (12)
According to General Procedure A, chlorin 11 (70 mg, 0.11 mmol) was reacted with NaBH4 (28 mg, 0.74 mmol) in 9:1 CH2Cl2/MeOH (3 mL) over 3 h in ambient conditions. Column chromatography (silica–3:1 CH2Cl2/hexane, then 99:1 CH2Cl2/ethyl acetate), followed by recrystallization of the main fraction (from CH2Cl2/MeOH), provided chlorin 12 as a violet solid in 50% yield (35 mg).
12: 1H NMR (500 MHz, CDCl3): δ = –1.81 (s, 1 H, NH), –1.78 (s, 1 H, NH), 0.93–1.00 (m, 12 H, 4 × CH3), 1.37–1.55 (m, 16 H, 8 × CH2), 1.70–1.83 (m, 8 H, 4 × CH2), 2.02–2.07 (m, 1 H, β-OH), 2.14–2.22 (m, 2 H, CH2), 2.24–2.32 (m, 2 H, CH2), 2.39–2.48 (m, 4 H, 2 × CH2), 4.03–4.20 (m, 2 H, CH2), 4.28–4.39 (m, 2 H, CH2), 4.40–4.52 (m, 2 H, β-H), 4.67–4.76 (m, 4 H, 2 × CH2), 6.58–6.63 (m, 1 H, β-H), 8.95 (d, J = 5.0 Hz, 1 H, β-H), 8.99 (d, J = 4.8 Hz, 1 H, β-H), 9.16 (d, J = 4.5 Hz, 1 H, β-H), 9.19 (d, J = 4.5 Hz, 1 H, β-H), 9.25–9.28 (m, 2 H, β-H), ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.35 (CH3), 22.92 (CH2), 22.98 (CH2), 30.33 (CH2), 30.43 (CH2), 30.47 (CH2), 30.52 (CH2), 32.07 (CH2), 32.09 (CH2), 34.12 (CH2), 35.01 (CH2), 35.23 (CH2), 35.43 (CH2), 35.57 (CH2), 37.20 (CH2), 38.18 (CH2), 38.23 (CH2), 44.50 (β-C-8), 73.24 (β-C-7), 109.93 (meso-C), 110.95 (meso-C), 120.80 (β-C), 121.06 (β-C), 121.77 (meso-C), 122.24 (meso-C), 124.90 (β-C), 125.53 (β-C), 129.58 (β-C), 130.02 (β-C), 134.21 (α-C), 134.81 (α-C), 139.71 (α-C), 139.82 (α-C), 151.81 (α-C), 152.71 (α-C), 161.44 (α-C), 162.69 (α-C) ppm. UV–vis (CH2Cl2): λmax (log ε): 410 (5.37), 430 (5.22), 529 (4.36), 556 (4.42), 596 (4.09), 649 (4.37) nm. HRMS (ESI): m/z calc’d for C44H65N4O+ ([M + H]+): 665.5153; found: 665.5156.
3.8. CTAP Oxidation of meso-Tetrahexylporphyrin (7) or meso-Tetrahexyl-7,8-cis-dihydroxychlorin (8): Formation of meso-Tetrahexylporpholactone (meso-Tetrahexyl-7-oxo-8-oxa-porphyrin) (13)
Prepared according to General Procedure B from porphyrin 7 or chlorin diol 8 (50 mg, 0.08 mmol, 1 equiv.) in CH2Cl2 (12 mL) and CTAP (0.375 mmol, 152 mg, 5 equiv.) over a 10–30 min reaction time. The crude mixture was chromatographed (silica–10% CH2Cl2/hexanes), resulting in a 76% yield of 13 (254 mg). Rf (silica–30% CH2Cl2/hexanes) = 0.54. 1H NMR (400 MHz, CD2Cl2): δ = 9.38 (s, 2 H), 6.38 (m, 2 H), 5.73-–5.66 (m, 4 H), 3.83 (d, J = 7.6 Hz, 1 H), 3.71 (m, 1 H), 3.65 (dt, J = 17.5, 8.9 Hz, 1 H), 3.65 (m, 4 H), 2.94 (dt, J = 17.3, 8.9 Hz, 1 H) ppm. {1H}13C NMR (101 MHz, CD2Cl2): δ = 117.1, 107.8.6, 107.4, 106.4, 78.5, 70.1, 67.2, 66.4, 40.5 ppm. UV–vis (CH2Cl2) λmax = 402 (Soret), 519, 547, 592, 634 nm. Fl (λexcitation = λSoret) λmax (rel. intensity) = 635 (1.0), 704 (0.17) nm. FT-IR spectrum (neat, ATR): νC=O = 1842 cm–1. HR-MS ESI+ (100% CH3CN, 30 V cone voltage): m/z = 664.4716 calc’d for C43H60N4O2 [M]+; found 664.4704.
3.9. DMP Oxidation of meso-Tetrahexylporphyrin (7): Formation of 5-(1′-oxo-hexyl)-10,15,20-trihexylporphyrin (14)
According to General Procedure C, porphyrin 7 (150 mg, 0.23 mmol) was reacted in 2:1 CH2Cl2/CH3CN (14 mL) with a 15% (w/w) solution of Dess–Martin periodinane (DMP) in CH2Cl2 (3.0 g, 3.6 mmol) over 12 h in ambient conditions. Column chromatography on silica–2:1 CH2Cl2/hexane. Recrystallization from CH2Cl2/MeOH provided product 14 as a purple solid in 44% yield (67 mg).
14: 1H NMR (500 MHz, CDCl3): δ = –2.72 (s, 2 H, NH), 0.93–0.97 (m, 12 H, 4 × CH3), 1.36–1.59 (m, 16 H, 8 × CH2), 1.76–1.85 (m, 6 H, 3 × CH2), 2.14 (m, 2 H, CH2), 2.44–2.56 (m, 6 H, 3 × CH2), 3.72 (t, J = 7.6 Hz, 2 H, CH2), 4.88 (t, J = 8.1 Hz, 4 H, 2 × CH2), 4.94 (t, J = 8.2 Hz, 2 H, CH2), 9.10 (d, J = 4.8 Hz, 2 H, β-H), 9.44 (d, J = 4.9 Hz, 2 H, β-H), 9.46 (d, J = 4.8 Hz, 2 H, β-H), 9.49 (d, J = 4.9 Hz, 2 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.14 (CH3), 14.30 (CH3), 14.31 (CH3), 22.76 (CH2), 22.89 (CH2), 22.91 (CH2), 25.45 (CH2), 30.39 (CH2), 30.48 (CH2), 31.81 (CH2), 32.05 (CH2), 32.06 (CH2), 35.51 (CH2), 36.10 (CH2), 38.83 (CH2), 39.10 (CH2), 50.79 (COCH2), 117.12 (meso-C), 120.05 (meso-C), 121.53 (meso-C), 210.22 (CO) ppm. UV–vis (CH2Cl2): λmax (log ε): 411 (5.52), 519 (4.19), 553 (3.86), 597 (3.65), 654 (3.68) nm. HRMS (ESI): m/z calc’d for C44H61N4O+ ([M + H]+): 661.4840; found: 661.4837.
3.10. CTAP Oxidation of 5,15-Dihexylporphyrin (15): Formation of 5,15-Dihexyl-3-oxo-2-oxa-porphyrin) (16A) and 5,15-Dihexyl-7-oxo-8-oxa-porphyrin) (16B)
Prepared according to General Procedure B from porphyrin 15 (50 mg, 0.1 mmol) in CH2Cl2 (12 mL) and CTAP (0.5 mmol, 202 mg, 5 equiv.) over a 5–10 min reaction time. The filtered reaction mixture was separated by preparative thin-layer chromatography (silica–40% CH2Cl2/hexanes) to obtain 16A in 55% yield (29 mg) 16B in less than 5% yield (<3 mg).
16A: Rf (silica–40% CH2Cl2/hexanes) = 0.67. 1H NMR (400 MHz, CD2Cl2): δ = –2.93 (s, 1 H), –2.23 (s, 1 H), 0.96 (t, J = 8.2 Hz, 6 H), 1.52–1.39 (m, 8 H), 1.74 (dd, J = 15.2, 7.7 Hz, 2 H), 1.82 (t, J = 7.5 Hz, 2 H), 2.34 (dq, J = 15.4, 7.7 Hz, 3 H), 2.50 (dt, J = 15.5, 7.8 Hz, 2 H), 4.52 (t, J = 7.9 Hz, 2 H), 4.87 (t, J = 8.0 Hz, 2 H), 9.10 (d, J = 4.4 Hz, 1 H), 9.19 (d, J = 3.4 Hz, 1 H), 9.30 (s, 2 H), 9.39 (d, J = 4.4 Hz, 1 H), 9.43 (d, J = 4.4 Hz, 1 H), 9.84 (s, 1 H), 9.93 (s, 1 H) ppm. 13C NMR (101 MHz; CD2Cl2): δ = 155.09, 154.92, 153.7, 140.8, 137.2, 136.3, 135.9, 134.9, 131.7, 131.0, 129.6, 126.1, 124.6, 124.3, 105.6, 101.8, 100.0, 38.4, 35.2, 34.9, 30.3, 29.80, 29.69 ppm. UV–vis (CH2Cl2) λmax (log ε) = 402 (Soret, 5.50), 509 (4.18), 546 (4.20), 577 (3.92), 584 (sh), 634 (3.95) nm. Fl (λexcitation = λSoret) λmax (rel. intensity) = 636 (1.0), 704 (0.18) nm. HR-MS ESI+ (100% CH3CN, 30 V cone voltage, TOF detection): m/z calc’d for C31H36N4O2 [M]+ 496.2838; found 496.2845.
16B: Rf (silica–40% CH2Cl2/hexanes) = 0.61. UV–vis (CH2Cl2) λmax (rel. I) = 402 (Soret, 1.0), 509 (0.047), 545 (0.050), 577 (0.026), 584 (sh), 633 (0.028) nm. MS ESI+ (100% CH3OH, 30 V cone voltage, TOF detection): m/z calc’d for C31H37N4O2 [M + H]+ 497.2911; found 497.2900.
3.11. Hydride reduction of meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-7,8-trans-dihydroxychlorin (17)
Prepared according to General Procedure A from dione 10 (25 mg, 0.03 mmol) in 95:5 CH2Cl2/MeOH (3 mL) and NaBH4 (10 mg, 0.26 mmol) over 90 min at 0 °C. Chromatographic purification of the crude material (silica–95:5 CH2Cl2/ethyl acetate) provided chlorin 17 in 52% yield (13 mg) as a purple solid.
17: 1H NMR (250 MHz, CDCl3): δ = –1.91 (br s, 2 H, NH), 0.90–0.99 (m, 12 H, 4 × CH3), 1.33–1.57 (m, 16 H, 8 × CH2), 1.69–1.85 (m, 8 H, 4 × CH2), 2.22–2.47 (m, 8 H, 4 × CH2), 4.24–4.34 (m, 4 H, 2 × CH2), 4.63–4.73 (m, 4 H, 2 × CH2), 6.26 (s, 2 H, β-H), 8.48 (d, J = 4.9 Hz, 2 H, β-H), 9.17 (s, 2 H, β-H), 9.26 (d, J = 4.9 Hz, 2 H, β-H) ppm. UV–vis (CH2Cl2): λmax (log ε): 409 (5.48), 426 sh (5.36), 528 (4.46), 558 (4.53), 594 (4.25), 648 (4.35) nm. HRMS (ESI): m/z calc’d for C44H65N4O2+ ([M + H]+): 681.5102; found: 681.5091.
3.12. Methyl-Grignard Addition to meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-8-hydroxy-8-methyl-chlorin-7-one (18) and meso-Tetrahexyl-7,8-dihydroxy-7,8-dimethyl-chlorin (19)
Dione 10 (50 mg, 0.07 mmol) was dissolved in dry THF (3 mL) and a 1 M solution of MeMgBr in THF (4 mL, 4 mmol) was added at –45 °C under inert conditions; the mixture was allowed to warm to ambient temperature and stirred for 3 d. After this time, water was added and the product mixture extracted with CH2Cl2 or ethyl acetate. The combined organic phases were dried over Na2SO4 (anhydr.) and chromatographed (silica–2:1 CH2Cl2/hexane); the first fraction of 18 was in a 24% (14 mg) and the second fraction of 19 in 19% (10 mg) yield, both as purple solids, after their crystallization from CH2Cl2/MeOH. Adding only 2 mL (2 mmol) of the 1 M MeMgBr solution in THF to dione 10 (50 mg, 0.07 mmol), dissolved in dry THF (3 mL) for a 5 h reaction time at –45 °C, followed by the same workup and chromatography conditions, yields only product 18 in 66% (34 mg) yield (66%)
18: 1H NMR (500 MHz, CDCl3): δ = –1.89 (s, 1 H, NH) –1.69 (s, 1 H, NH), 0.92–0.98 (m, 12 H, 4 × CH3), 1.35–1.53 (m, 16 H, 8 × CH2), 1.71–1.84 (m, 8 H, 4 × CH2), 1.94 (s, 3 H, β-CH3), 1.96–2.28 (m, 4 H, 2 × CH2), 2.38–2.45 (m, 4 H, 2 × CH2), 3.56 (s, 1 H, β-OH), 4.48–4.55 (m, 1 H, CHA), 4.62–4.78 (m, 7 H, 3 × CH2, CHB), 9.15 (d, J = 4.7 Hz, 1 H, β-H), 9.17–9.19 (m, 1 H, β-H), 9.22 (d, J = 4.7 Hz, 1 H, β-H), 9.23–9.24 (m, 1 H, β-H), 9.28–9.31 (m, 2 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.32 (CH3), 22.90 (CH2), 22.99 (CH2), 26.07 (β-CH3), 30.25 (CH2), 30.37 (CH2), 30.43 (CH2), 30.72 (CH2), 31.50 (CH2), 32.04 (CH2), 32.60 (CH2), 35.38 (CH2), 35.54 (CH2), 36.47 (CH2), 37.76 (CH2), 38.29 (CH2), 81.22 (C-7), 111.80 (meso-C), 115.95 (meso-C), 120.57 (meso-C), 123.29 (β-C), 124.68 (β-C), 124.70 (β-C), 126.17 (β-C), 130.68 (β-C), 131.43 (β-C), 136.66 (α-C), 136.33 (α-C), 138.22 (α-C), 140.39 (α-C), 140.65 (α-C), 152.68 (α-C), 154.53 (α-C), 159.27 (α-C), 212.62 (β-CO) ppm. UV–vis (CH2Cl2): λmax (log ε): 421 (5.17), 441 (5.07), 541 (4.05), 578 (4.12), 607 (3.90), 662 (3.72) nm. HRMS (ESI): m/z calc’d for C45H65N4O2+ ([M + H]+): 693.5102; found: 693.5081.
19: 1H NMR (500 MHz, CDCl3): δ = –1.10 (s, 2 H, NH), 0.91–0.97 (m, 12 H, 4 × CH3), 1.34–1.50 (m, 16 H, 8 × CH2), 1.66–1.76 (m, 14 H, 4 × CH2, 2 × β-CH3), 1.93–2.01 (m, 2 H, CH2), 2.06–2.16 (m, 2 H, CH2), 2.33–2.40 (m, 4 H, 2 × CH2), 2.88 (s, 2 H, β-OH), 4.26–4.34 (m, 2 H, CH2), 4.46–4.53 (m, 2 H, CH2), 4.56–4.66 (m, 4 H, 2 × CH2), 8.95 (d, J = 4.9 Hz, 2 H, β-H), 9.07 (s, 2 H, β-H), 9.17 (d, J = 4.9 Hz, 2 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.31 (CH3), 22.89 (CH2), 23.00 (CH2), 23.57 (β-CH3), 30.40 (CH2), 30.68 (CH2), 31.68 (CH2), 32.04 (CH2), 35.18 (CH2), 37.79 (CH2), 37.91 (CH2), 90.27 (β-C), 111.52 (meso-C), 121.99 (meso-C), 122.06 (β-C), 125.39 (β-C), 129.60 (β-C), 134.13 (α-C), 141.19 (α-C), 151.72 (α-C), 159.47 (α-C) ppm. UV–vis (CH2Cl2): λmax (log ε): 415 (4.95), 436 (4.75), 535 (4.03), 565 (4.08), 602 (3.85), 657 (3.99) nm. HRMS (ESI): m/z calc’d for C46H69N4O2+ ([M + H]+): 709.5415; found: 709.5432.
3.13. Trifluoromethylation of meso-Tetrahexylporphyrin-7,8-dione (10): Formation of meso-Tetrahexyl-8-hydroxy-8-trifluoromethyl-chlorin-7-one (18F)
Dione 10 was dissolved in THF (3 mL) and trifluoromethyltrimethylsilane (50 µL, 0.38 mmol) was added between –35 and –45 °C, in addition to a catalytic quantity of tetrabutylammonium fluoride (TBAF). After 20 min of stirring, additional TBAF (33 mg, 0.11 mmol) was added and the mixture stirred for 10 min. The mixture was allowed to warm, water was added, and the product extracted with CH2Cl2 or ethyl acetate. The combined organic fractions were dried over Na2SO4 (anhydr.), the solvent removed using rotary evaporation, and the residue chromatographed (silica–1:1 CH2Cl2/hexane). Recrystallization of the main fraction from CH2Cl2/MeOH provided 18F in 73% yield (40 mg) as a purple solid.
18F: 1H NMR (500 MHz, CDCl3): δ = –1.76 (s, 1 H, NH), –1.62 (s, 1 H, NH), 0.91–1.00 (m, 12 H, 4 × CH3), 1.35–1.53 (m, 16 H, 8 × CH2), 1.69–1.86 (m, 8 H, 4 × CH2), 1.99–2.15 (m, 3 H, CH2, CHA), 2.25–2.35 (m, 1 H, CHB), 2.36–2.46 (m, 4 H, 2 × CH2), 4.45–4.57 (m, 4 H, CH2, CHA, 1 × β-OH), 4.65–4.77 (m, 5 H, 4 × CH2, CHB), 9.15–9.17 (m, 1 H, β-H), 9.19–9.21 (m, 2 H, β-H), 9.22–9.24 (m, 1 H, β-H), 9.30–9.33 (m, 2 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.30 (CH3), 14.34 (CH3), 22.89 (CH2), 22.99 (CH2), 30.15 (CH2), 30.37 (CH2), 30.42 (CH2), 30.70 (CH2), 31.72 (CH2), 31.97 (CH2), 32.02 (CH2), 33.22 (CH2), 35.52 (CH2), 36.31 (CH2), 38.10 (CH2), 38.31 (CH2), 38.34 (CH2), 113.41 (meso-C), 115.09 (meso-C),122.10 (meso-C), 123.88 (β-C), 124.97 (β-C), 125.26 (β-C), 126.22 (β-C), 131.12 (β-C), 131.63 (β-C), 136.09 (α-C), 136.36 (α-C), 138.36 (α-C), 139.88 (α-C), 140.39 (α-C), 149.11 (α-C), 153.34 (α-C), 154.60 (α-C), 205.89 (β-CO) ppm. 19F NMR (471 MHz, CDCl3): δ = –75.36 (s, 3 F, CF3) ppm. UV–vis (CH2Cl2): λmax (log ε): 426 (5.16), 449 (5.12), 552 (4.10), 588 (4.19), 612 (4.13), 668 (3.99) nm. HRMS (ESI): m/z calc’d for C45H62F3N4O2+ ([M + H]+): 747.4819; found: 747.4822.
3.14. Hydride Reduction of meso-Tetrahexyl-8-hydroxy-8-methyl-chlorin-7-one (18): Formation of meso-Tetrahexyl-7,8-dihydroxy-8-methyl-chlorin (20)
According to General Procedure A, meso-tetrahexyl-7,8-dihydroxy-8-methyl-chlorin 20 was obtained from meso-tetrahexyl-8-hydroxy-8-methyl-chlorin-7-one (18) (25 mg, 0.04 mmol) in 9:1 CH2Cl2 (3 mL) at 0 °C and NaBH4 (5 mg, 0.13 mmol) over 90 min. Chromatographic purification (silica–95:5 CH2Cl2/ethyl acetate) and recrystallization from CH2Cl2/ethyl MeOH provided 20 in 80% yield (20 mg) as a purple solid.
20: 1H NMR (500 MHz, CDCl3): δ = –1.73 (s, 2 H, NH), 0.91–0.98 (m, 12 H, 4 × CH3), 1.35–1.52 (m, 16 H, 8 × CH2), 1.64–1.79 (m, 8 H, 4 × CH2), 2.07–2.23 (m, 7 H, 2 × CH2, 1 × β-CH3), 2.36–2.45 (m, 4 H, 2 × CH2), 4.26–4.45 (m, 3 H, CH2, CHA), 4.51–4.74 (m, 5 H, 2 × CH2, CHB), 6.38 (s, 1 H, β-H), 8.98 (d, J = 5.0 Hz, 1 H, β-H), 9.01 (d, J = 4.9 Hz, 1 H, β-H), 9.11–9.14 (m, 2 H, β-H), 9.21 (d, J = 5.0 Hz, 1 H, β-H), 9.23 (d, J = 4.9 Hz, 1 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ = 14.32 (CH3), 22.52 (β-CH3), 22.91 (CH2), 22.95 (CH2), 22.98 (CH2), 30.40 (CH2), 30.44 (CH2), 30.54 (CH2), 32.05 (CH2), 32.54 (CH2), 33.95 (CH2), 35.22 (CH2), 35.53 (CH2), 36.79 (CH2), 37.51 (CH2), 38.07 (CH2), 38.17 (CH2), 85.32 (β-C), 87.37 (β-C), 111.04 (meso-C), 111.31 (meso-C), 121.54 (meso-C), 121.92 (meso-C), 121.15 (β-C), 122.40 (β-C), 125.19 (β-C), 125.21 (β-C), 129.80 (β-C), 130.01 (β-C), 134.28 (α-C), 134.74 (α-C), 140.05 (α-C), 140.86 (α-C), 152.11 (α-C), 152.43 (α-C), 158.06 (α-C), 161.69 (α-C) ppm. UV–vis (CH2Cl2): λmax (log ε): 410 (5.18), 431 (5.02), 531 (4.09), 559 (4.21), 598 (3.88), 652 (4.07) nm. HRMS (ESI+, TOF detection): m/z calc’d for C45H67N4O2+ ([M + H]+): 695.5259; found: 695.5240.
3.15. Hydride Reduction of meso-Tetrahexyl-8-hydroxy-8-trifluoromethyl-chlorin-7-one (18F): meso-Tetrahexyl-7,8-dihydroxy-8-trifluoromethyl-chlorin (20F)
According to General Procedure A, meso-tetrahexyl-7,8-dihydroxy-8-trifluoromethyl-chlorin 20F was obtained from meso-tetrahexyl-8-hydroxy-8-trifluoromethyl-chlorin-7-one (18F) (25 mg, 0.03 mmol) in 9:1 CH2Cl2 (3 mL) at 0 °C and NaBH4 (5 mg, 0.13 mmol) over 90 min. Chromatographic purification (silica–95:5 CH2Cl2/ethyl acetate) and recrystallization from CH2Cl2/MeOH provided 20F in 80% yield (21 mg) as a purple solid.
20F: 1H NMR (500 MHz, CDCl3): δ = –1.51 (br s, 2 H, NH), 0.88–0.99 (m, 12 H, 4 × CH3), 1.26–1.61 (m, 18 H, 9 × CH2), 1.71–1.82 (m, 6 H, 3 × CH2), 1.95–2.02 (m, 2 H, CH2), 2.16–2.33 (m, 2 H, CH2), 2.36–2.44 (m, 4 H, 2 × CH2), 2.84–2.93 (m, 1 H, β-OH), 3.93 (br s, 1 H, β-OH), 4.20–4.28 (m, 1 H, CHA), 4.35–4.46 (m, 3 H, CH2, CHB), 4.56–4.72 (m, 4 H, 2 × CH2), 6.93–6.96 (m, 1 H, β-H), 8.97 (d, J = 5.1 Hz, 1 H, β-H), 9.03 (d, J = 5.0 Hz, 1 H, β-H), 9.09 (d, J = 4.6 Hz, 1 H, β-H), 9.13 (d, J = 4.6 Hz, 1 H, β-H), 9.20–9.22 (m, 2 H, β-H) ppm. {1H}13C NMR (126 MHz, CDCl3): δ =14.25 (CH3), 14.30 (CH3), 22.89 (CH2), 23.00 (CH2), 30.21 (CH2), 30.39 (CH2), 30.44 (CH2), 30.62 (CH2), 31.96 (CH2), 32.04 (CH2), 32.87 (CH2), 33.04 (CH2), 35.12 (CH2), 35.55 (CH2), 36.47 (CH2), 38.04 (CH2), 38.09 (CH2), 38.15 (CH2), 89.18 (β-C), 109.89 (meso-C), 113.78 (meso-C), 121.49 (meso-C), 122.52 (β-C), 122.85 (β-C), 123.30 (meso-C), 124.95 (β-C), 125.80 (β-C), 129.88 (β-C), 130.41 (β-C), 134.24 (α-C), 135.02 (α-C), 140.30 (α-C), 140.58 (α-C), 149.30 (α-C), 153.15 (α-C), 153.25 (α-C), 155.25 (α-C) ppm. {1H}19F NMR (471 MHz, CDCl3): δ = –72.65 (s, 3 F, CF3) ppm. UV–vis (CH2Cl2): λmax (log ε): 409 (5.34), 431 (5.17), 533 (4.22), 562 (4.40), 599 (4.08), 653 (4.24) nm. HRMS (ESI): m/z calc’d for C45H64F3N4O2+ ([M + H]+): 749.4976; found: 749.4955.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29092144/s1: reproductions of the key spectra of all new compounds.
Author Contributions
Conceptualization, C.B.W.S., A.W. and C.B.; methodology, investigation, and data curation, D.A. and D.D.; writing—original draft preparation, C.B.; writing—review and editing, all authors; supervision, project administration, and funding acquisition, C.B.W.S., A.W. and C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the US National Science Foundation under grant number CHE-1800361 (to C.B.). The support of this research by the Arbeitsgemeinschaft industrieller Forschungsvereinigungen ‘Otto-von-Guericke’ e. V. (AiF) (Projects KF0579901UL7 and KF0249303UL7) (to A.W.) is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Scheer, H. An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions, and Applications. In Chlorophylls and Bacteriochlorophylls; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 1–26. [Google Scholar]
- Liu, Y.; Zhang, S.; Lindsey, J.S. Total synthesis campaigns toward chlorophylls and related natural hydroporphyrins–diverse macrocycles, unrealized opportunities. Nat. Prod. Rep. 2018, 35, 879–901. [Google Scholar] [CrossRef] [PubMed]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, S.; Wang, X.; Qian, Y.; Wu, W.; Zhang, S.; Zheng, B.; Wei, G.; Gao, S.; Cao, Z.; et al. High Affinity of Chlorin e6 to Immunoglobulin G for Intraoperative Fluorescence Image-Guided Cancer Photodynamic and Checkpoint Blockade Therapy. ACS Nano 2019, 13, 10242–10260. [Google Scholar] [CrossRef] [PubMed]
- Sutton, P.A.; van Dam, M.A.; Cahill, R.A.; Mieog, S.; Polom, K.; Vahrmeijer, A.L.; van der Vorst, J. Fluorescence-guided surgery: Comprehensive review. BJS Open 2023, 7, zrad049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.; Zeng, Z.; Pu, K. Activatable molecular probes for fluorescence-guided surgery, endoscopy and tissue biopsy. Chem. Soc. Rev. 2022, 51, 566–593. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K. Synthetic strategies in designing porphyrin-based photosensitizers for photodynamic therapy. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004; pp. 144/1–144/21. [Google Scholar]
- Zhi, D.; Yang, T.; O’Hagan, J.; Zhang, S.; Donnelly, R.F. Photothermal therapy. J. Control. Release 2020, 325, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Photoantimicrobials—So what’s stopping us? Photodiagn. Photodyn. Ther. 2009, 6, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, A.; O’Brien, J.M.; Senge, M.O. Trends and targets in antiviral phototherapy. Photochem. Photobiol. Sci. 2019, 18, 2565–2612. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.K.; Ladomenou, K.; Coutsolelos, A.G. Porphyrins in bio-inspired transformations: Light-harvesting to solar cell. Coord. Chem. Rev. 2012, 256, 2601–2627. [Google Scholar] [CrossRef]
- Jiang, J.; Matula, A.J.; Swierk, J.R.; Romano, N.; Wu, Y.; Batista, V.S.; Crabtree, R.H.; Lindsey, J.S.; Wang, H.; Brudvig, G.W. Unusual Stability of a Bacteriochlorin Electrocatalyst under Reductive Conditions. A Case Study on CO2 Conversion to CO. ACS Catal. 2018, 8, 10131–10136. [Google Scholar] [CrossRef]
- Brückner, C.; Samankumara, L.; Ogikubo, J. Syntheses of Bacteriochlorins and Isobacteriochlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2012; Volume 17, pp. 1–112. [Google Scholar]
- Lindsey, J.S. De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating Our Green World. Chem. Rev. 2015, 115, 6534–6620. [Google Scholar] [CrossRef] [PubMed]
- Borbas, K.E. Chlorins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NY, USA, 2016; Volume 36, pp. 1–149. [Google Scholar]
- Taniguchi, M.; Lindsey, J.S. Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins. Chem. Rev. 2017, 117, 344–535. [Google Scholar] [CrossRef] [PubMed]
- Jux, N.; Montforts, F.-P.; Haake, E. Bacteriochlorins and Isobacteriochlorins (Tetrahydroporphyrins), and Hexahydroporphyrins. In Science of Synthesis Knowledge Updates; Georg Thieme Verlag KG: Stuttgart, Germany, 2022; pp. 111–141. [Google Scholar] [CrossRef]
- Fischer, H.; Eckoldt, H. Überführung von Porphyrinen in Dioxy-chlorine durch Einwirkung von Osmiumtetroxyd. Liebigs Ann. Chem. 1940, 544, 138–162. [Google Scholar] [CrossRef]
- Pandey, R.K.; Isaac, M.; MacDonald, I.; Medforth, C.J.; Senge, M.O.; Dougherty, T.J.; Smith, K.M. Pinacol-Pinacolone Rearrangements in vic-Dihydroxychlorins and Bacteriochlorins: Effect of Substituents at the Peripheral Positions. J. Org. Chem. 1997, 62, 1463–1472. [Google Scholar] [CrossRef]
- Adams, K.R.; Bonnett, R.; Burke, P.J.; Salgado, A.; Valles, M.A. Cleavage of (octaethyl-2,3-dihydroxychlorinato)nickel(II) to give the novel 2,3-dioxo-2,3-secochlorin system. J. Chem. Soc. Perkin Trans. 1 1997, 1769–1772. [Google Scholar] [CrossRef]
- Brückner, C.; Rettig, S.J.; Dolphin, D. Formation of a meso-Tetraphenylsecochlorin and a Homoporphyrin with a Twist. J. Org. Chem. 1998, 63, 2094–2098. [Google Scholar] [CrossRef]
- Sutton, J.M.; Fernandez, N.; Boyle, R.W. Functionalized diphenylchlorins and bacteriochlorins: Their synthesis and bioconjugation for targeted photodynamic therapy and tumour cell imaging. J. Porphyr. Phthalocyanines 2000, 4, 655–658. [Google Scholar] [CrossRef]
- Rancan, F.; Wiehe, A.; Nöbel, M.; Senge, M.O.; Omari, S.A.; Böhm, F.; John, M.; Röder, B. Influence of substitutions on asymmetric dihydroxychlorins with regard to intracellular uptake, subcellular localization and photosensitization of Jurkat cells. J. Photochem. Photobiol. B Biol. 2005, 78, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Hyland, M.A.; Morton, M.D.; Brückner, C. meso-Tetra(pentafluorophenyl)porphyrin-derived Chromene-annulated Chlorins. J. Org. Chem. 2012, 77, 3038–3048. [Google Scholar] [CrossRef]
- Bruhn, T.; Brückner, C. Origin of the Regioselective Reduction of Chlorins. J. Org. Chem. 2015, 80, 4861–4868. [Google Scholar] [CrossRef]
- Lalisse, R.F.; Hadad, C.M.; Brückner, C.; Guberman-Pfeffer, M.J. [3 + 2]-Cycloadditions with Porphyrin β,β′-Bonds: Theoretical Basis of the Counterintuitive meso-Aryl Group Influence on the Rates of Reaction. J. Org. Chem. 2022, 87, 16473–16482. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Sotiriou, C. Migratory aptitudes in pinacol rearrangement of vic-dihydroxychlorins. J. Heterocycl. Chem. 1985, 22, 1739–1741. [Google Scholar] [CrossRef]
- Starnes, S.D.; Rudkevich, D.M.; Rebek Jr., J. Cavitand-Porphyrins. J. Am. Chem. Soc. 2001, 123, 4659–4669. [Google Scholar] [CrossRef] [PubMed]
- Brückner, C. The Breaking and Mending of meso-Tetraarylporphyrins: Transmuting the Pyrrolic Building Blocks. Acc. Chem. Res. 2016, 49, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.P.; Atoyebi, A.O.; Tardie, W.; Zeller, M.; Brückner, C. Pyrrole-Modified Porphyrins Containing Eight-Membered Heterocycles Using a Reversal of the “Breaking and Mending” Strategy. J. Org. Chem. 2020, 85, 15273–15286. [Google Scholar] [CrossRef] [PubMed]
- Egorov, G.D.; Solov’ev, V.N.; Shul’ga, A.M. PMR spectra of symmetrical meso-substituted porphyrins and chlorine. Theor. Exp. Chem. 1975, 11, 77–86. [Google Scholar]
- Ulman, A.; Gallicci, J.; Fisher, D.; Ibers, J.A. Facile Synthesis of Tetraalkylchlorin and Tetraalkylporphyrin Complexes and Comparison to the Structures of the Tetramethylchlorin and Tetramethylporphyrin Complexes of Ni(II). J. Am. Chem. Soc. 1980, 102, 6852–6854. [Google Scholar] [CrossRef]
- Frydman, L.; Olivieri, A.C.; Diaz, L.E.; Frydman, B.; Morin, F.G.; Mayne, C.L.; Grant, D.M.; Adler, A.D. High-Resolution Solid-State 13C NMR Spectra of Porphine and 5,10,15,20-Tetraalkylporphyrins: Implications for the N-H Tautomerization Process. J. Am. Chem. Soc. 1988, 110, 336–342. [Google Scholar] [CrossRef]
- Geier, G.R.; Lindsey, J.S. Effects of aldehyde or dipyrromethane substituents on the reaction course leading to meso-substituted porphyrins. Tetrahedron 2004, 60, 11435–11444. [Google Scholar] [CrossRef]
- Smith, B.M.; Kean, S.D.; Wyatt, M.F.; Graham, A.E. Indium Triflate Mediated Synthesis of meso-Substituted Porphyrins. Synlett 2008, 1953–1956. [Google Scholar] [CrossRef]
- DiMagno, S.G.; Williams, R.A.; Therien, M.J. Facile Synthesis of meso-Tetrakis(perfluoroalkyl)porphyrins: Spectroscopic Properties and X-ray Crystal Structure of Highly Electron-Deficient 5,10,15,20-Tetrakis(heptafluoropropyl)porphyrin. J. Org. Chem. 1994, 59, 6943–6948. [Google Scholar] [CrossRef]
- Senge, M.O.; Bischoff, I.; Nelson, N.Y.; Smith, K.M. Synthesis, reactivity and structural chemistry of 5,10,15,20-tetraalkylporphyrins. J. Porphyr. Phthalocyanines 1999, 3, 99–116. [Google Scholar] [CrossRef]
- Feng, X.; Senge, M.O. One-pot synthesis of functionalized asymmetric 5,10,15,20-substituted porphyrins from 5,15-diaryl- or -dialkyl-porphyrins. Tetrahedron 2000, 56, 587–590. [Google Scholar] [CrossRef]
- Feng, X.; Senge, M.O. An efficient synthesis of highly functionalized asymmetric porphyrins with organolithium reagents. J. Chem. Soc. Perkin Trans. 1 2001, 1, 1030–1038. [Google Scholar] [CrossRef]
- Nam, D.T.; Ivanova, Y.B.; Puhovskaya, S.G.; Kruk, M.M.; Syrbu, S.A. Acid–base equilibria and coordination chemistry of the 5,10,15,20-tetraalkyl-porphyrins: Implications for metalloporphyrin synthesis. RSC Adv. 2015, 5, 26125–26131. [Google Scholar] [CrossRef]
- Hiroto, S.; Osuka, A. Meso-Alkyl-Substituted meso-meso Linked Diporphyrins and meso-Alkyl-Substituted meso-meso, b-b, b-b Triply Linked Diporphyrins. J. Org. Chem. 2005, 70, 4054–4058. [Google Scholar] [CrossRef]
- Paliteiro, C.; Sobral, A. Electrochemical and spectroelectrochemical characterization of meso-tetra-alkyl porphyrins. Electrochim. Acta 2005, 50, 2445–2451. [Google Scholar] [CrossRef]
- Wiehe, A.; Shaker, Y.M.; Brandt, J.C.; Mebs, S.; Senge, M.O. Lead structures for applications in photodynamic therapy. Part 1: Synthesis and variation of m-THPC (Temoporfin) related amphiphilic A2BC-type porphyrins. Tetrahedron 2005, 61, 5535–5564. [Google Scholar] [CrossRef]
- Dahms, K.; Senge, M.O.; Bakri Bakar, M. Exploration of meso-substituted formyl porphyrins and their Grignard and Wittig reactions. Eur. J. Org. Chem. 2007, 2007, 3833–3848. [Google Scholar] [CrossRef]
- Senge, M.O.; Shaker, Y.M.; Pintea, M.; Ryppa, C.; Hatscher, S.S.; Ryan, A.; Sergeeva, Y. Synthesis of meso-Substituted ABCD-Type Porphyrins by Functionalization Reactions. Eur. J. Org. Chem. 2010, 2010, 237–258. [Google Scholar] [CrossRef]
- Plamont, R.; Kikkawa, Y.; Takahashi, M.; Kanesato, M.; Giorgi, M.; Chan Kam Shun, A.; Roussel, C.; Balaban, T.S. Nanoscopic Imaging of meso-Tetraalkylporphyrins Prepared in High Yields Enabled by Montmorrilonite K10 and 3Å Molecular Sieves. Chem.–Eur. J. 2013, 19, 11293–11300. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Osuka, A.; Yamamoto, Y.; Tokuji, S.; Tanaka, T. Palladium-Catalyzed Tetraarylation of 5,15-Dialkylporphyrins with Aryl Bromides. Heterocycles 2014, 88, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Król, A.; Plamont, R.; Canard, G.; Edzang, J.A.; Gryko, D.T.; Balaban, T.S. An Efficient Synthesis of Porphyrins with Different meso Substituents that Avoids Scrambling in Aqueous Media. Chem.–Eur. J. 2015, 21, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.R.; Panduwinata, D.; Visser, J.; Chin, Y.; Tang, C.; Goerigk, L.; Ford, M.J.; Baker, M.; Sum, T.J.; Coenen, M.J.J.; et al. From Chaos to Order: Chain-Length Dependence of the Free Energy of Formation of meso-Tetraalkylporphyrin Self-Assembled Monolayer Polymorphs. J. Phys. Chem. C 2016, 120, 1739–1748. [Google Scholar] [CrossRef]
- Jiang, X.-L.; Damunupola, D.; Brückner, C. meso-Tetra(dioxanyl)porphyrins: Neutral, low molecular weight, and chiral porphyrins with solubility in aqueous solutions. J. Porphyr. Phthalocyanines 2021, 25, 734–740. [Google Scholar] [CrossRef]
- Aicher, D.; Wiehe, A.; Stark, C.B.W.; Albrecht, V.; Gräfe, S. Preparation of β-Functionalized Dihydroxy-Chlorins for Photodynamic Therapy. WO 2012012809, 26 January 2012. [Google Scholar]
- Aicher, D.; Wiehe, A.; Stark, C.B.W.; Albrecht, V.; Gräfe, S. Application of Beta-Functionalized Dihydroxy-Chlorins for PDT. U.S. Patent 20130041307, 24 April 2013. [Google Scholar]
- Gallucci, J.C.; Swepston, P.N.; Ibers, J.A. The Structures of (5,10,15,20-Tetramethylporphyrinato)nickel(II) and (5,10,15,20-Tetramethylchlorinato)nickel(II). Acta Cryst. Sect. B 1982, 38, 2134–2139. [Google Scholar] [CrossRef]
- Ulman, A.; Fisher, D.; Ibers, J.A. Synthesis of some 5,10,15,20-tetraalkylchlorin and tetraalkylporphyrin complexes of transition metals. J. Heterocycl. Chem. 1982, 19, 409–413. [Google Scholar] [CrossRef]
- Gouterman, M. Optical spectra and electronic structure of porphyrins and related rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 3, pp. 1–165. [Google Scholar]
- Richter, A.M.; Waterfield, E.; Jain, A.K.; Sternberg, E.D.; Dolphin, D.; Levy, J.G. In vitro evaluation of phototoxic properties of four structurally related benzoporphyrin derivatives. Photochem. Photobiol. 1990, 52, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Boyle, R.W.; Dolphin, D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem. Photobiol. 1996, 64, 469–485. [Google Scholar] [CrossRef]
- Samankumara, L.P.; Zeller, M.; Krause, J.A.; Brückner, C. Syntheses, structures, modification, and optical properties of meso-tetraaryl-2,3-dimethoxychlorin, and two isomeric meso-tetraaryl-2,3,12,13-tetrahydroxybacteriochlorins. Org. Biomol. Chem. 2010, 8, 1951–1965. [Google Scholar] [CrossRef]
- Hyland, M.A.; Hewage, N.; Walton, K.; Nimthong Roldan, A.; Zeller, M.; Samaraweera, M.; Gascon, J.A.; Brückner, C. Chromene-annulated Bacteriochlorins. J. Org. Chem. 2016, 81, 3603–3618. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.W.; Williams, S.C.; Jenkins, H.A.; Brückner, C. Oxidation of meso-tetraphenyl-2,3-dihydroxychlorin: Simplified synthesis of ß,ß’-dioxochlorins. Tetrahedron Lett. 2003, 44, 4045–4049. [Google Scholar] [CrossRef]
- Brückner, C.; Dolphin, D. 2,3-vic-Dihydroxy-meso-tetraphenylchlorins from the osmium tetroxide oxidation of meso-tetraphenylporphyrin. Tetrahedron Lett. 1995, 36, 3295–3298. [Google Scholar] [CrossRef]
- Dean, M.L.; Schmink, J.R.; Leadbeater, N.E.; Brückner, C. Microwave-promoted insertion of Group 10 metals into free base porphyrins and chlorins: Scope and limitations. Dalton Trans. 2008, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Medforth, C.J.; Smith, K.M.; Alderfer, J.; Dougherty, T.J.; Pandey, R.K. Effect of meso-Substituents on the Osmium Tetraoxide Reaction and Pinacol-Pinacolone Rearrangement of the Corresponding vic-Dihydroxyporphyrins. J. Org. Chem. 2001, 66, 3930–3939. [Google Scholar] [CrossRef]
- Crossley, M.J.; Harding, M.M.; Sternhell, S. Tautomerism in 2-substituted 5,10,15,20-tetraphenylporphyrins. J. Am. Chem. Soc. 1986, 108, 3608–3613. [Google Scholar] [CrossRef]
- Crossley, M.J.; Harding, M.M.; Sternhell, S. Tautomerism in 2-hydroxy-5,10,15,20-tetraphenylporphyrin: An equilibrium between enol, keto, and aromatic hydroxyl tautomers. J. Org. Chem. 1988, 53, 1132–1137. [Google Scholar] [CrossRef]
- Burns, D.H.; Li, Y.H.; Shi, D.C.; Delaney, M.O. C-H Bond activation by alumina: Facile hydroxylation of chlorins at their saturated b-carbon by molecular oxygen and alumina. Chem. Commun. 1998, 1677–1678. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Jenkins, H.A.; Brückner, C. Free Base meso-Tetraaryl-morpholinochlorins and Porpholactone from meso-Tetraaryl-2,3-dihydroxy-chlorin. Org. Lett. 2003, 5, 19–22. [Google Scholar] [CrossRef]
- Brückner, C.; Ogikubo, J.; McCarthy, J.R.; Akhigbe, J.; Hyland, M.A.; Daddario, P.; Worlinsky, J.L.; Zeller, M.; Engle, J.T.; Ziegler, C.J.; et al. Oxazolochlorins. 6. meso-Arylporpholactones and their Reduction Products. J. Org. Chem. 2012, 77, 6480–6494. [Google Scholar] [CrossRef]
- Hewage, N.; Daddario, P.; Lau, K.S.F.; Guberman-Pfeffer, M.J.; Gascón, J.A.; Zeller, M.; Lee, C.O.; Khalil, G.E.; Gouterman, M.; Brückner, C. Bacterio- and Isobacteriodilactones by Stepwise or Direct Oxidations of meso-Tetrakis(pentafluorophenyl)porphyrin. J. Org. Chem. 2019, 84, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Thuita, D.; Damunupola, D.; Brückner, C. Oxazolochlorins 21. Most Efficient Access to meso-Tetraphenyl- and meso-Tetrakis(pentafluorophenyl)porpholactones, and Their Zinc(II) and Platinum(II) Complexes. Molecules 2020, 25, 4351. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.J.; King, L.G. Novel heterocyclic systems from selective oxidation at the β-pyrrolic position of porphyrins. J. Chem. Soc. Chem. Commun. 1984, 920–922. [Google Scholar] [CrossRef]
- Gouterman, M.; Hall, R.J.; Khalil, G.E.; Martin, P.C.; Shankland, E.G.; Cerny, R.L. Tetrakis(pentafluorophenyl)porpholactone. J. Am. Chem. Soc. 1989, 111, 3702–3707. [Google Scholar] [CrossRef]
- Jayaraj, K.; Gold, A.; Austin, R.N.; Ball, L.M.; Terner, J.; Mandon, D.; Weiss, R.; Fischer, J.; DeCian, A.; Bill, E.; et al. Compound I and Compound II Analogues from Porpholactones. Inorg. Chem. 1997, 36, 4555–4566. [Google Scholar] [CrossRef] [PubMed]
- Köpke, T.; Pink, M.; Zaleski, J.M. Elucidation of the extraordinary 4-membered pyrrole ring-contracted azeteoporphyrinoid as an intermediate in chlorin oxidation. Chem. Commun. 2006, 4940–4942. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lv, H.; Ke, X.; Yang, B.; Zhang, J.-L. Ruthenium-Catalyzed Oxidation of the Porphyrin ß-ß’-Pyrrolic Ring: A General and Efficient Approach to Porpholactones. Adv. Synth. Catal. 2012, 354, 3509–3516. [Google Scholar] [CrossRef]
- Arnold, L.; Müllen, K. Modifying the Porphyrin Core—A Chemist’s Jigsaw. J. Porphyr. Phthalocyanines 2011, 15, 757–779. [Google Scholar] [CrossRef]
- Brückner, C.; Akhigbe, J.; Samankumara, L. Syntheses and Structures of Porphyrin Analogues Containing Non-pyrrolic Heterocycles. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: River Edge, NJ, USA, 2014; Volume 31, pp. 1–276. [Google Scholar]
- Thuita, D.W.; Brückner, C. Metal Complexes of Porphyrinoids Containing Nonpyrrolic Heterocycles. Chem. Rev. 2022, 122, 7990–8052. [Google Scholar] [CrossRef]
- Heravi, M.M.; Momeni, T.; Zadsirjan, V.; Mohammadi, L. Applications of the Dess-Martin Oxidation in Total Synthesis of Natural Products. Curr. Org. Synth. 2021, 18, 125–196. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Jothish, S.; Dohi, T.; Singh, F.V. Iodine(V)-Based Oxidants in Oxidation Reactions. Molecules 2023, 28, 5250. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Jin, G.-Q.; Zhang, J.-L. Porpholactone Chemistry: An Emerging Approach to Bioinspired Photosensitizers with Tunable Near-Infrared Photophysical Properties. Acc. Chem. Res. 2019, 52, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Hewage, N.; Damunupola, D.; Zeller, M.; Brückner, C. Direct Oxidations of meso-Tetrakis(pentafluorophenyl)porphyrin: Porphotrilactones and Entry into a Non-biological Porphyrin Degradation Pathway. J. Org. Chem. 2024, 89, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.J.; Burn, P.L.; Langford, S.J.; Pyke, S.M.; Stark, A.G. A New Method for the Synthesis of Porphryin-alpha-diones that is Applicable to the Synthesis of Trans-annular Extended Porphyrin Systems. J. Chem. Soc. Chem. Commun. 1991, 1567–1568. [Google Scholar] [CrossRef]
- Akhigbe, J.; Brückner, C. Expansion of a Pyrrole in meso-Tetraphenylporphyrin to a Pyrazine Imide Moiety Using a Beckmann Rearrangement. Eur. J. Org. Chem. 2013, 2013, 3876–3884. [Google Scholar] [CrossRef]
- Banerjee, S.; Zeller, M.; Brückner, C. meso-Tetraphenylporphyrin-derived Oxypyriporphyrin, Oxypyrichlorin, and Thiomorpholinochlorin, as their Ni(II) Complexes. J. Porphyr. Phthalocyanines 2012, 16, 576–588. [Google Scholar] [CrossRef]
- Ruppert, I.; Schlich, K.; Volbach, W. Die ersten CF3-substituierten organyl(chlor)silane. Tetrahedron Lett. 1984, 25, 2195–2198. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Krishnamurti, R.; Olah, G.A. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc. 1989, 111, 393–395. [Google Scholar] [CrossRef]
- Hewage, N.; Zeller, M.; Brückner, C. Oxidations of chromene-annulated chlorins. Org. Biomol. Chem. 2017, 15, 396–407. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, MA, USA, 2009. [Google Scholar]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman: Essex, UK, 1989; p. 549. [Google Scholar]
Scheme 1.
The osmium tetroxide-mediated dihydroxylation of β-alkyl- (1) and meso-arylporphyrins (3) to generate the corresponding dihydroxychlorins 2 and 4, respectively.
Scheme 1.
The osmium tetroxide-mediated dihydroxylation of β-alkyl- (1) and meso-arylporphyrins (3) to generate the corresponding dihydroxychlorins 2 and 4, respectively.

Figure 1.
General structure of meso-tetraalkylporphyrins 5 and literature-known [meso-tetramethylchlorinato]nickel(II) (6).
Figure 1.
General structure of meso-tetraalkylporphyrins 5 and literature-known [meso-tetramethylchlorinato]nickel(II) (6).

Scheme 2.
Dihydroxylation of meso-tetrahexylporphyrin 7 to produce the corresponding dihydroxychlorin 8 and tetrahydroxybacteriochlorin 9.
Scheme 2.
Dihydroxylation of meso-tetrahexylporphyrin 7 to produce the corresponding dihydroxychlorin 8 and tetrahydroxybacteriochlorin 9.

Figure 2.
UV–vis spectrum of the compounds indicated (CH2Cl2).

Scheme 3.
Functional group transformations of meso-tetrahexyl-2,3-dihydroxychlorin 8.

Figure 3.
Stacked, normalized UV–vis spectra of the compounds indicated (CH2Cl2).

Scheme 4.
One-step oxidative transformations of meso-tetrahexylporphyrin 7.

Scheme 5.
Direct CTAP oxidation of meso-5,15-dihexylporphyrin 15.

Figure 4.
Illustrated partial 1H,13C-HMBC NMR spectrum observed for dihexylporpholactone isomer 16A.
Figure 4.
Illustrated partial 1H,13C-HMBC NMR spectrum observed for dihexylporpholactone isomer 16A.

Scheme 6.
Transformations of meso-tetrahexylporphyrin-7,8-dione 10.

Figure 5.
UV–vis spectra of the compounds indicated (CH2Cl2).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aicher, D.; Damunupola, D.; Stark, C.B.W.; Wiehe, A.; Brückner, C. meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives. Molecules 2024, 29, 2144. https://doi.org/10.3390/molecules29092144
AMA Style
Aicher D, Damunupola D, Stark CBW, Wiehe A, Brückner C. meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives. Molecules. 2024; 29(9):2144. https://doi.org/10.3390/molecules29092144
Chicago/Turabian StyleAicher, Daniel, Dinusha Damunupola, Christian B. W. Stark, Arno Wiehe, and Christian Brückner. 2024. "meso-Tetrahexyl-7,8-dihydroxychlorin and Its Conversion to ß-Modified Derivatives" Molecules 29, no. 9: 2144. https://doi.org/10.3390/molecules29092144