
Development of a Robust Read-Across Model for the Prediction of Biological Potency of Novel Peroxisome Proliferator-Activated Receptor Delta Agonists
 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Interpretation of the Selected Descriptors
2.2. Model Validation
2.2.1. Metrics and Statistics
2.2.2. Internal and External Validation
2.3. Applicability Domain
2.4. Model Availability
- ]
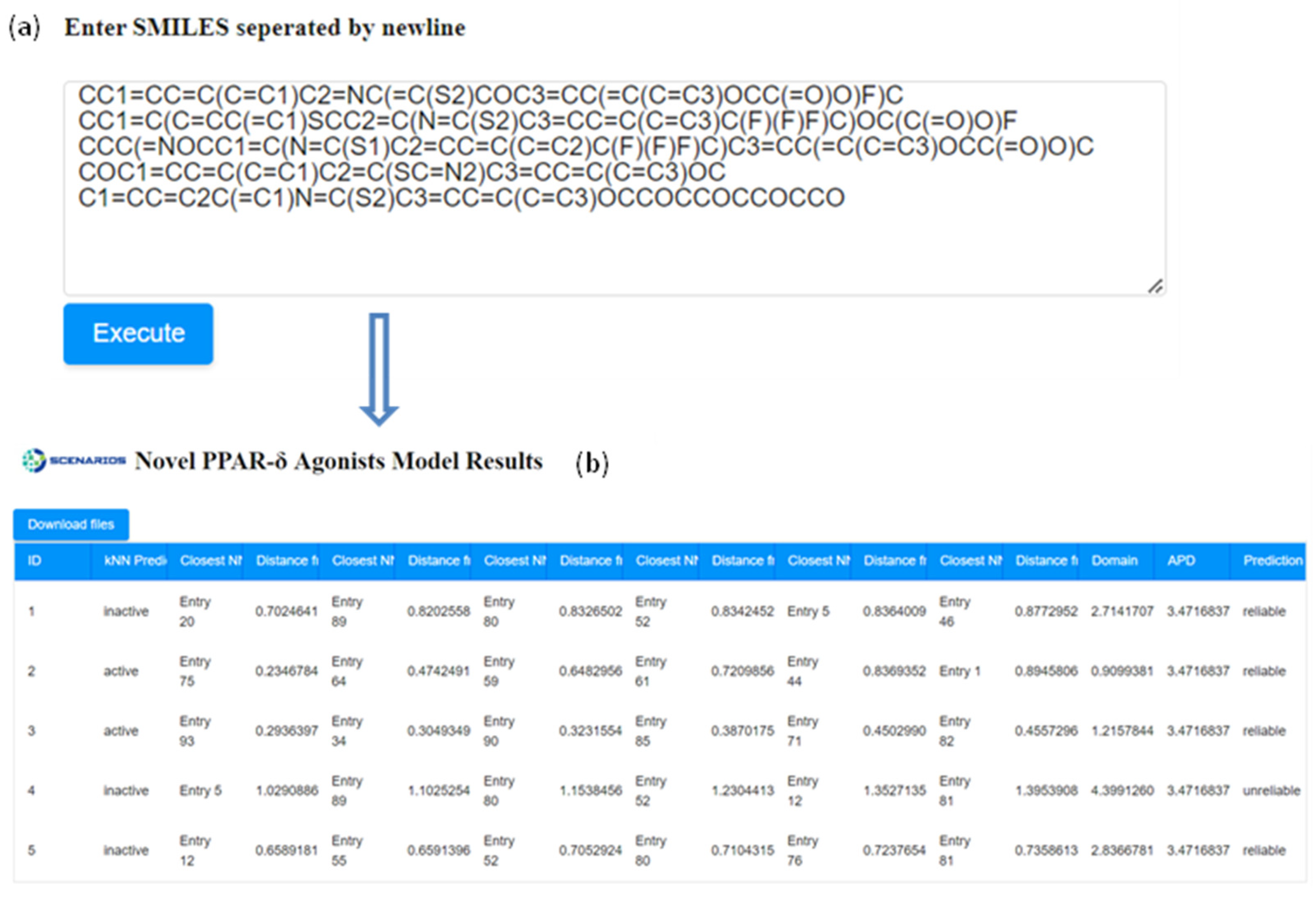
- The user is able to make a request through a data transfer software such as Client URL:
- curl -X POST “https://enaloscloud.novamechanics.com/scenarios/apis/ppardelta/smiles” -H “accept: application/json” -H “Content-Type: application/json” -d “[ { \”smiles\”: \”Cc1c(ccc(c1)OCc2nc(c(o2)-c3ccc(cc3)OC(F)(F)F)-c4cnccc4)OCC(=O)O\” }]”
- and obtain the corresponding results of the GUI environment, as seen in Figure 4. The returned response includes class prediction, the closest neighbours, and the Euclidean distances from the molecule in question, and the APD indicating the reliability of the prediction:
- [
- {
- “id”: “cluster_0”,
- “idNN1”: “Entry 20”,
- “distNN1”: 0.7024641202204505,
- “idNN2”: “Entry 89”,
- “distNN2”: 0.8202558963827292,
- “idNN3”: “Entry 80”,
- “distNN3”: 0.8326502973790398,
- “idNN4”: “Entry 52”,
- “distNN4”: 0.8342452655891001,
- “idNN5”: “Entry 5”,
- “distNN5”: 0.8364009145055961,
- “idNN6”: “Entry 46”,
- “distNN6”: 0.8772952879358424,
- “domain”: 2.714170703054797,
- “apd”: 3.4716837408236625,
- “predictionReliablility”: “reliable”,
- “knnprediction”: “inactive”
- }
- ]
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Calculation of Descriptors
4.3. Data Pre-Processing
4.4. Clustering into Distinct Classes
4.5. Variable Selection
4.6. Molecular Docking Calculations
4.7. Model Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | Accuracy |
| APD | Applicability domain |
| API | Application Programming Interfaces |
| CoMFA | Comparative molecular field analysis and |
| CoMSIA | Comparative molecular similarity indices analysis |
| DBD | DNA-binding domain |
| EC50 | Half maximal effective concentration |
| FN | False negative |
| FP | False positive |
| GUI | Graphical user interface |
| HQSAR | Hologram quantitative structure–activity relationships |
| ISE | Iterative stochastic elimination |
| MCC | Matthews correlation coefficient |
| NIH | National Institutes of Health |
| NR | Nuclear receptor |
| OECD | Organisation for Economic Co-operation and Development |
| PFAS | Per- and polyfluoroalkyl substances |
| PPAR | Peroxisome proliferator-activated receptor |
| PPRE | Peroxisome proliferator response element |
| QMRF | QSAR model reporting format |
| QSAR | Quantitative structure–activity relationship |
| REST | Representational state transfer |
| RXR | Retinoid X receptor |
| S2k | Kier shape index (fixed length k = 2) |
| SDF | Structure-data file |
| SMILES | Simplified molecular input line entry system |
| TN | True negative |
| TP | True positive |
| kNN | k-nearest neighbours |
| κ | Cohen’s kappa |
References
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The Nuclear Receptor Superfamily: A Structural Perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Sharma, S.; Gupta, P.; Saini, A.; Kaushal, C. The Peroxisome Proliferator-Activated Receptor: A Family of Nuclear Receptors Role in Various Diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236. [Google Scholar] [CrossRef]
- Georgiadi, A.; Kersten, S. Mechanisms of Gene Regulation by Fatty Acids. Adv. Nutr. 2012, 3, 127–134. [Google Scholar] [CrossRef]
- Ferré, P. The Biology of Peroxisome Proliferator-Activated Receptors. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Martin, G.; Staels, B.; Auwerx, J. Peroxisome Proliterator-Activated Receptors, Orphans with Ligands and Functions. Curr. Opin. Lipidol. 1997, 8, 159–166. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome Proliferator-Activated Receptors: Nuclear Control of Metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Palioura, D.; Mellidis, K.; Mouchtouri, E.-T.; Mavroidis, M.; Lazou, A. PPARβ/δ at the Crossroads of Energy Metabolism, Mitochondrial Quality Control and Redox Balance. J. Biol. Res.-Thessalon. 2022, 29, 12. [Google Scholar] [CrossRef]
- Abuhammad, A.; Taha, M.O. QSAR Studies in the Discovery of Novel Type-II Diabetic Therapies. Expert Opin. Drug Discov. 2016, 11, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Lather, V.; Fernandes, M.X. QSAR Models for Prediction of PPARδ Agonistic Activity of Indanylacetic Acid Derivatives. QSAR Comb. Sci. 2009, 28, 447–457. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Homem-de-Mello, P.; Honorio, K.M. Role of Physicochemical Properties in the Activation of Peroxisome Proliferator-Activated Receptor δ. J. Mol. Model. 2011, 17, 2549–2558. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Silva, D.C.; Honório, K.M. Advanced QSAR Studies on PPARδ Ligands Related to Metabolic Diseases. J. Braz. Chem. Soc. 2012, 23, 78–84. [Google Scholar] [CrossRef]
- Wickens, P.; Zhang, C.; Ma, X.; Zhao, Q.; Amatruda, J.; Bullock, W.; Burns, M.; Cantin, L.-D.; Chuang, C.-Y.; Claus, T.; et al. Indanylacetic Acids as PPAR-δ Activator Insulin Sensitizers. Bioorganic Med. Chem. Lett. 2007, 17, 4369–4373. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.S.; Silva, D.C.; Gertrudes, J.C.; Maltarollo, V.G.; Honorio, K.M. Molecular Features Related to the Binding Mode of PPAR δ Agonists from QSAR and Docking Analyses. SAR QSAR Environ. Res. 2013, 24, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Ding, T.-T.; Feng, X.-Y.; Xu, W.-R.; Cheng, X.-C. Virtual Identification of Novel Peroxisome Proliferator-Activated Receptor (PPAR) α/δ Dual Antagonist by 3D-QSAR, Molecule Docking, and Molecule Dynamics Simulation. J. Biomol. Struct. Dyn. 2020, 38, 4143–4161. [Google Scholar] [CrossRef] [PubMed]
- Maltarollo, V.G.; Araujo, S.C.; Trossini, G.H.G.; Honorio, K.M. Understanding PPAR-δ Affinity and Selectivity Using Hologram Quantitative Structure–Activity Modeling, Molecular Docking and GRID Calculations. Future Med. Chem. 2016, 8, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.S.; Honório, K.M. Two-Dimensional Quantitative Structure-Activity Relationship Studies on Bioactive Ligands of Peroxisome Proliferator-Activated Receptor δ. J. Braz. Chem. Soc. 2011, 22, 65–72. [Google Scholar] [CrossRef]
- Shearer, B.G.; Patel, H.S.; Billin, A.N.; Way, J.M.; Winegar, D.A.; Lambert, M.H.; Xu, R.X.; Leesnitzer, L.M.; Merrihew, R.V.; Huet, S.; et al. Discovery of a Novel Class of PPARδ Partial Agonists. Bioorganic Med. Chem. Lett. 2008, 18, 5018–5022. [Google Scholar] [CrossRef] [PubMed]
- Da’adoosh, B.; Marcus, D.; Rayan, A.; King, F.; Che, J.; Goldblum, A. Discovering Highly Selective and Diverse PPAR-Delta Agonists by Ligand Based Machine Learning and Structural Modeling. Sci. Rep. 2019, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Kadayat, T.M.; Shrestha, A.; Jeon, Y.H.; An, H.; Kim, J.; Cho, S.J.; Chin, J. Targeting Peroxisome Proliferator-Activated Receptor Delta (PPARδ): A Medicinal Chemistry Perspective. J. Med. Chem. 2020, 63, 10109–10134. [Google Scholar] [CrossRef]
- OECD. Reconciling Terminology of the Universe of Per- and Polyfluoroalkyl Substances: Recommendations and Practical Guidance; OECD Series on Risk Management; No. 61; OECD Publishing: Paris, France, 2021; Available online: https://www.oecd.org/chemicalsafety/portal-perfluorinated-chemicals/terminology-per-and-polyfluoroalkyl-substances.pdf (accessed on 4 December 2023).
- European Chemicals Agency. Read-Across Assessment Framework (RAAF); European Chemicals Agency: Helsinki, Finland, 2017. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley: Hoboken, NJ, USA, 2000; ISBN 9783527299133. [Google Scholar]
- Moreau, G.; Broto, P. The Auto-Correlation of a Topological-Structure—A New Molecular Descriptor. New J. Chem. 1980, 4, 359–360. [Google Scholar]
- Broto, P.; Moreau, G.; Vandycke, C. Molecular Structures–Perception, Auto-Correlation Descriptor and SAR Studies -Autocorrelation Descriptor. Eur. J. Med. Chem. 1984, 19, 66–70. [Google Scholar]
- Burden, F.R. A Chemically Intuitive Molecular Index Based on the Eigenvalues of a Modified Adjacency Matrix. Quant. Struct.-Act. Relatsh. 1997, 16, 309–314. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Kier, L.B. Shape Indexes of Orders One and Three from Molecular Graphs. Quant. Struct.-Act. Relatsh. 1986, 5, 1–7. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular Recognition of Fatty Acids by Peroxisome Proliferator–Activated Receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Naser, M.Z.; Alavi, A.H. Error Metrics and Performance Fitness Indicators for Artificial Intelligence and Machine Learning in Engineering and Sciences. Archit. Struct. Constr. 2021, 3, 499–517. [Google Scholar] [CrossRef]
- Faulon, J.-L.; Bender, A. Handbook of Chemoinformatics Algorithms; Chapman and Hall/CRC: Boca Raton, FL, USA, 2010; ISBN 9781420082999. [Google Scholar]
- NovaMechanics Ltd. Enalos + KNIME Nodes. 2017. Available online: http://enalosplus.novamechanics.com/ (accessed on 4 December 2023).
- Afantitis, A.; Melagraki, G.; Koutentis, P.A.; Sarimveis, H.; Kollias, G. Ligand-Based Virtual Screening Procedure for the Prediction and the Identification of Novel β-Amyloid Aggregation Inhibitors Using Kohonen Maps and Counterpropagation Artificial Neural Networks. Eur. J. Med. Chem. 2011, 46, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Melagraki, G.; Afantitis, A.; Sarimveis, H.; Igglessi-Markopoulou, O.; Koutentis, P.A.; Kollias, G. In Silico Exploration for Identifying Structure–Activity Relationship of MEK Inhibition and Oral Bioavailability for Isothiazole Derivatives. Chem. Biol. Drug Des. 2010, 76, 397–406. [Google Scholar] [CrossRef]
- Varsou, D.D.; Melagraki, G.; Sarimveis, H.; Afantitis, A. MouseTox: An Online Toxicity Assessment Tool for Small Molecules through Enalos Cloud Platform. Food Chem. Toxicol. 2017, 110, 83–93. [Google Scholar] [CrossRef]
- Melagraki, G.; Ntougkos, E.; Rinotas, V.; Papaneophytou, C.; Leonis, G.; Mavromoustakos, T.; Kontopidis, G.; Douni, E.; Afantitis, A.; Kollias, G. Cheminformatics-Aided Discovery of Small-Molecule Protein-Protein Interaction (PPI) Dual Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-ΚB Ligand (RANKL). PLoS Comput. Biol. 2017, 13, e1005372. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current Clinical Trial Status and Future Prospects of PPAR-Targeted Drugs for Treating Nonalcoholic Fatty Liver Disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef] [PubMed]
- Ramaprasad, A.S.E.; Smith, M.T.; McCoy, D.; Hubbard, A.E.; La Merrill, M.A.; Durkin, K.A. Predicting the Binding of Small Molecules to Nuclear Receptors Using Machine Learning. Brief. Bioinform. 2022, 23, bbac114. [Google Scholar] [CrossRef] [PubMed]
- Varsou, D.-D.; Tsoumanis, A.; Papadiamantis, A.G.; Melagraki, G.; Afantitis, A. Isalos Predictive Analytics Platform: Cheminformatics, Nanoinformatics, and Data Mining Applications. In Machine Learning and Deep Learning in Computational Toxicology; Springer: Cham, Switzerland, 2023; pp. 223–242. [Google Scholar]
- Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD: Paris, France, 2014; ISBN 9789264085442.
- European Commission; Joint Research Centre (JRC). JRC QSAR Model Database; European Commission, Joint Research Centre (JRC) [Dataset] PID; Joint Research Centre (JRC): Brussels, Belgium, 2020; Available online: http://data.europa.eu/89h/E4ef8d13-D743-4524-A6eb-80e18b58cba4 (accessed on 4 December 2023).
- Epple, R.; Cow, C.; Xie, Y.; Azimioara, M.; Russo, R.; Wang, X.; Wityak, J.; Karanewsky, D.S.; Tuntland, T.; Nguyêñ-Trân, V.T.B.; et al. Novel Bisaryl Substituted Thiazoles and Oxazoles as Highly Potent and Selective Peroxisome Proliferator-Activated Receptor δ Agonists. J. Med. Chem. 2010, 53, 77–105. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Bioassay Record for AID 469785, Source: ChEMBL. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/469785 (accessed on 4 December 2023).
- Singh, A.; Raju, R.; Mrad, M.; Reddell, P.; Münch, G. The Reciprocal EC50 Value as a Convenient Measure of the Potency of a Compound in Bioactivity-Guided Purification of Natural Products. Fitoterapia 2020, 143, 104598. [Google Scholar] [CrossRef] [PubMed]
- Nandy, A.; Roy, K.; Saha, A. Exploring Molecular Fingerprints of Selective PPARδ Agonists through Comparative and Validated Chemometric Techniques. SAR QSAR Environ. Res. 2015, 26, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, Molecular Descriptors from 2D Structures for Chemoinformatics and Toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. J. R. Stat. Soc. Ser. C (Appl. Stat.) 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Witten, I.H.; Frank, E.; Hall, M.A.; Pal, C.J. Data Mining; Morgan Kaufmann: Burlington, MA, USA, 2017. [Google Scholar]
- West, R.M. Best Practice in Statistics: The Use of Log Transformation. Ann. Clin. Biochem. Int. J. Lab. Med. 2022, 59, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Daszykowski, M.; Walczak, B.; Massart, D.L. Representative Subset Selection. Anal. Chim. Acta 2002, 468, 91–103. [Google Scholar] [CrossRef]
- Tang, S.; Chen, R.; Lin, M.; Lin, Q.; Zhu, Y.; Ding, J.; Hu, H.; Ling, M.; Wu, J. Accelerating AutoDock Vina with GPUs. Molecules 2022, 27, 3041. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.H.; Trivella, D.B.B.; Bernardes, A.; Gratieri, J.; Oliveira, P.S.L.; Figueira, A.C.M.; Webb, P.; Polikarpov, I. Structural Insights into Human Peroxisome Proliferator Activated Receptor Delta (PPAR-Delta) Selective Ligand Binding. PLoS ONE 2012, 7, e33643. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, D.; Drakopoulos, A.; Lagarias, P.; Melagraki, G.; Kollias, G.; Afantitis, A. In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes. Int. J. Mol. Sci. 2021, 22, 10220. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. Amber 2021: Reference Manual; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Cover, T.; Hart, P. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Varsou, D.; Afantitis, A.; Tsoumanis, A.; Papadiamantis, A.; Valsami-Jones, E.; Lynch, I.; Melagraki, G. Zeta-Potential Read-Across Model Utilizing Nanodescriptors Extracted via the NanoXtract Image Analysis Tool Available on the Enalos Nanoinformatics Cloud Platform. Small 2020, 16, 1906588. [Google Scholar] [CrossRef]
- Varsou, D.D.; Ellis, L.J.A.; Afantitis, A.; Melagraki, G.; Lynch, I. Ecotoxicological Read-across Models for Predicting Acute Toxicity of Freshly Dispersed versus Medium-Aged NMs to Daphnia Magna. Chemosphere 2021, 285, 131452. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Predicted Active | Predicted Inactive |
|---|---|---|
| Actual Active | 20 | 3 |
| Actual Inactive | 2 | 16 |
| Metric | Metric Formula | Metric Value |
|---|---|---|
| Accuracy | 87.8% | |
| Sensitivity | 87.0% | |
| Precision | 90.9% | |
| F1-Score | 88.9% | |
| Matthews Correlation Coefficient | 0.755 | |
| Cohen’s kappa | 0.754 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoniou, M.; Papavasileiou, K.D.; Melagraki, G.; Dondero, F.; Lynch, I.; Afantitis, A. Development of a Robust Read-Across Model for the Prediction of Biological Potency of Novel Peroxisome Proliferator-Activated Receptor Delta Agonists. Int. J. Mol. Sci. 2024, 25, 5216. https://doi.org/10.3390/ijms25105216
Antoniou M, Papavasileiou KD, Melagraki G, Dondero F, Lynch I, Afantitis A. Development of a Robust Read-Across Model for the Prediction of Biological Potency of Novel Peroxisome Proliferator-Activated Receptor Delta Agonists. International Journal of Molecular Sciences. 2024; 25(10):5216. https://doi.org/10.3390/ijms25105216
Chicago/Turabian StyleAntoniou, Maria, Konstantinos D. Papavasileiou, Georgia Melagraki, Francesco Dondero, Iseult Lynch, and Antreas Afantitis. 2024. "Development of a Robust Read-Across Model for the Prediction of Biological Potency of Novel Peroxisome Proliferator-Activated Receptor Delta Agonists" International Journal of Molecular Sciences 25, no. 10: 5216. https://doi.org/10.3390/ijms25105216