
Recent Advances in the Discovery of SIRT1/2 Inhibitors via Computational Methods: A Perspective
 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Structural Information of SIRT1-2
2.1. X-ray Crystallographic Structures of SIRT1
2.2. X-ray Crystallographic Structures of SIRT2
3. Computational Methods toward the Discovery of SIRT1-2 Modulators
3.1. Design of SIRT1 Modulators via Computational Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R.D. | Ref. | Type of Computational Method | Selectivity over Other Isoforms | Screened Database (n. of Compounds) | Most Active Compound/Proposed Compound | Activator/ Inhibitor | Potency |
|---|---|---|---|---|---|---|---|---|
| 1 | 2008 | [67] | Structure-based–Pharmacophore-based | 1,2 | The Maybridge and Leadquest libraries |  Oxadiazole-carbonylaminothiourea | I | 13 μM (IC50, SIRT1) |
| 2 | 2011 | [69] | Sequence-based VS | Not tested | SPECS drug-like library (85,000) |  Various | I | 5.72 μM (IC50) |
| 3 | 2012 | [71] | Target: HM | Not tested | In-house database (2500) |  Acridinedione derivatives | I | 0.25 μM (IC50) |
| 4 | 2014 | [72] | Target model for inhibitors: crystal structure; Target model for activators: HM of the allosteric site | Not tested | Asinex (>600,000) |  | I | 16.35 μM (IC50) |
| 5 | 2016 | [73] | SBVS | Not tested | Drug bank library from ZINC (1716) |  Diphenyl and oxycoumarin derivatives | I | 77.7% inhibition at 5 μM |
| 6 | 2020 | [74] | Iterative in vitro and in silico screenings | 2,3,5 | Small library of previously identified putative SIRT-1 inhibitors | 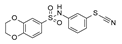 Thiocyanates | I | 5.2 μM (IC50) |
| 7 | 2021 | [75] | SBVS | Not tested | In-house library (54) |  | A | 79% at 100 μM |
| 8 | 2022 | [76] | SBVS | Not tested | In-house library of 1,8-dioxo-octahydroxanthene derivatives (18) | 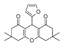 | I | 87.6% at 10 μM |
| 9 | 2022 | [77] | SBVS | 2,3 | In-house library (1,000,000) |  | I | 0.66 μM |
| 10 | 2023 | [78] | SBVS | 2,3 | SPECS database |  hsa62 | I | 1.3 μM (IC50) |
| 11 | 2016 | [79] | LB and SB pharmacophore models combined with docking-based VS | Not tested | ASINEX (5 000 000), in-house (971) | 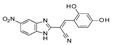 Benzimidazole derivative | I | 4.34 μM (IC50) |
| 12 | 2019 | [80] | LB and SB pharmacophore models | Not tested | Indonesian Herbal Database (1377) |  Mulberrin, quinine, quinidine, and gartanin | A | 1.14 μM (EC50) |
| 13 | 2019 | [81] | Contest-based approach (combined methods) | Not tested | ENAMINE (2 459 912) |  various various | I | 4.1 μM (IC50) |
| 14 | 2022 | [82] | LB and SB methods | Not tested | In-house library |  | A | 62.3% at 100 μM |
3.2. Design of SIRT2 Inhibitors via Computational Approaches
3.3. Design of Dual SIRT1-2 Inhibitors via Computational Approaches
4. Recently Exploited Chemical Scaffolds in the Search for SIRT1/2 Inhibitors
4.1. Design of SIRT1Is
4.2. Design of SIRT2Is
4.3. Design of Dual SIRT1/2Is
4.4. Design of Pan SIRTIs
5. Conclusions
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fiorentino, F.; Mautone, N.; Menna, M.; D’Acunzo, F.; Mai, A.; Rotili, D. Sirtuin Modulators: Past, Present, and Future Perspectives. Future Med. Chem. 2022, 14, 915–939. [Google Scholar] [CrossRef] [PubMed]
- Mautone, N.; Zwergel, C.; Mai, A.; Rotili, D. Sirtuin Modulators: Where Are We Now? A Review of Patents from 2015 to 2019. Expert Opin. Ther. Pat. 2020, 30, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Nandave, M.; Acharjee, R.; Bhaduri, K.; Upadhyay, J.; Rupanagunta, G.P.; Ansari, M.N. A Pharmacological Review on SIRT 1 and SIRT 2 Proteins, Activators, and Inhibitors: Call for Further Research. Int. J. Biol. Macromol. 2023, 242, 124581. [Google Scholar] [CrossRef] [PubMed]
- Lambona, C.; Zwergel, C.; Valente, S.; Mai, A. SIRT3 Activation a Promise in Drug Development? New Insights into SIRT3 Biology and Its Implications on the Drug Discovery Process. J. Med. Chem. 2024, 67, 1662–1689. [Google Scholar] [CrossRef] [PubMed]
- Baran, M.; Miziak, P.; Stepulak, A.; Cybulski, M. The Role of Sirtuin 6 in the Deacetylation of Histone Proteins as a Factor in the Progression of Neoplastic Disease. Int. J. Mol. Sci. 2023, 25, 497. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C. Sirtuin 6—A Key Regulator of Hepatic Lipid Metabolism and Liver Health. Cells 2023, 12, 663. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, C.; Tan, J.; Dou, C.; Chen, Y. Modulation of SIRT6 Activity Acts as an Emerging Therapeutic Implication for Pathological Disorders in the Skeletal System. Genes Dis. 2023, 10, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, Y.-Y.; Li, L.-S.; Liu, Y.-S. Sirtuins at the Crossroads between Mitochondrial Quality Control and Neurodegenerative Diseases: Structure, Regulation, Modifications, and Modulators. Aging Dis. 2023, 14, 794. [Google Scholar] [CrossRef] [PubMed]
- Abbotto, E.; Scarano, N.; Piacente, F.; Millo, E.; Cichero, E.; Bruzzone, S. Virtual Screening in the Identification of Sirtuins’ Activity Modulators. Molecules 2022, 27, 5641. [Google Scholar] [CrossRef]
- Manjula, R.; Anuja, K.; Alcain, F.J. SIRT1 and SIRT2 Activity Control in Neurodegenerative Diseases. Front. Pharmacol. 2021, 11, 585821. [Google Scholar] [CrossRef]
- de Oliveira, R.M.; Vicente Miranda, H.; Francelle, L.; Pinho, R.; Szegö, É.M.; Martinho, R.; Munari, F.; Lázaro, D.F.; Moniot, S.; Guerreiro, P.; et al. The Mechanism of Sirtuin 2–Mediated Exacerbation of Alpha-Synuclein Toxicity in Models of Parkinson Disease. PLoS Biol. 2017, 15, e2000374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Hong, T.-T.; Sun, Y.; Huang, H.; Chen, F.; Chen, X.; Chen, H.; Dong, S.; Cui, L.; et al. RTN4B-mediated Suppression of Sirtuin 2 Activity Ameliorates Β-amyloid Pathology and Cognitive Impairment in Alzheimer’s Disease Mouse Model. Aging Cell 2020, 19, e13194. [Google Scholar] [CrossRef]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.L.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S.; et al. Salermide, a Sirtuin Inhibitor with a Strong Cancer-Specific Proapoptotic Effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.; Uciechowska, U.; Schemies, J.; Rumpf, T.; Jung, M.; Sippl, W. Inhibitors to Understand Molecular Mechanisms of NAD+-Dependent Deacetylases (Sirtuins). Biochim. Biophys. Acta (BBA)–Gene Regul. Mech. 2010, 1799, 726–739. [Google Scholar] [CrossRef]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; Depinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor Activity of a Small-Molecule Inhibitor of Human Silent Information Regulator 2 Enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef]
- Voogd, T.E.; Vansterkenburg, E.L.; Wilting, J.; Janssen, L.H. Recent Research on the Biological Activity of Suramin. Pharmacol. Rev. 1993, 45, 177–203. [Google Scholar]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in Vivo Activity, and Mechanism of Action of a Small-Molecule P53 Activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue Alpha-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.G.; Kim, W.; Choi, M.; Kim, J.-E. AK-1, a Specific SIRT2 Inhibitor, Induces Cell Cycle Arrest by Downregulating Snail in HCT116 Human Colon Carcinoma Cells. Cancer Lett. 2015, 356, 637–645. [Google Scholar] [CrossRef]
- Mellini, P.; Itoh, Y.; Tsumoto, H.; Li, Y.; Suzuki, M.; Tokuda, N.; Kakizawa, T.; Miura, Y.; Takeuchi, J.; Lahtela-Kakkonen, M.; et al. Potent Mechanism-Based Sirtuin-2-Selective Inhibition by an in Situ-Generated Occupant of the Substrate-Binding Site, “Selectivity Pocket” and NAD+ Binding Site. Chem. Sci. 2017, 8, 6400–6408. [Google Scholar] [CrossRef]
- Schiedel, M.; Robaa, D.; Rumpf, T.; Sippl, W.; Jung, M. The Current State of NAD+ -Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. Med. Res. Rev. 2018, 38, 147–200. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An Exploratory Double-Blind, Randomized Clinical Trial with Selisistat, a SirT1 Inhibitor, in Patients with Huntington’s Disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef]
- Pérez-Peña, H.; Abel, A.-C.; Shevelev, M.; Prota, A.E.; Pieraccini, S.; Horvath, D. Computational Approaches to the Rational Design of Tubulin-Targeting Agents. Biomolecules 2023, 13, 285. [Google Scholar] [CrossRef]
- Dorahy, G.; Chen, J.Z.; Balle, T. Computer-Aided Drug Design towards New Psychotropic and Neurological Drugs. Molecules 2023, 28, 1324. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Bruzzone, S.; Nencioni, A.; Del Rio, A. Selectivity Hot-Spots of Sirtuin Catalytic Cores. Mol. Biosyst. 2015, 11, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Gerhardt, S.; Einsle, O.; Jung, M. Seeding for Sirtuins: Microseed Matrix Seeding to Obtain Crystals of Human Sirt3 and Sirt2 Suitable for Soaking. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.M.; Huber, F.M.; Hoelz, A. Structural and Functional Analysis of Human SIRT1. J. Mol. Biol. 2014, 426, 526–541. [Google Scholar] [CrossRef]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal Structure Analysis of Human Sirt2 and Its ADP-Ribose Complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal Structures of Human SIRT3 Displaying Substrate-Induced Conformational Changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Gertz, M.; Nguyen, G.T.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A Molecular Mechanism for Direct Sirtuin Activation by Resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB—5X16: Sirt6 Apo Structure. Available online: https://www.rcsb.org/structure/5X16 (accessed on 27 July 2023).
- Cao, D.; Wang, M.; Qiu, X.; Liu, D.; Jiang, H.; Yang, N.; Xu, R.-M. Structural Basis for Allosteric, Substrate-Dependent Stimulation of SIRT1 Activity by Resveratrol. Genes. Dev. 2015, 29, 1316–1325. [Google Scholar] [CrossRef]
- RCSB PDB—4IF6: Structure of NAD-Dependent Protein Deacetylase Sirtuin-1 (Closed State, 2.25 A). Available online: https://www.rcsb.org/structure/4IF6 (accessed on 31 July 2023).
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 Å Crystal Structure of the SIRT1 Catalytic Domain Bound to Nicotinamide Adenine Dinucleotide (NAD+) and an Indole (EX527 Analogue) Reveals a Novel Mechanism of Histone Deacetylase Inhibition. J. Med. Chem. 2013, 56, 963–969, Erratum in J. Med. Chem. 2016, 59, 2267. [Google Scholar] [CrossRef]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic Structure of a Small Molecule SIRT1 Activator-Enzyme Complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef] [PubMed]
- Disch, J.S.; Evindar, G.; Chiu, C.H.; Blum, C.A.; Dai, H.; Jin, L.; Schuman, E.; Lind, K.E.; Belyanskaya, S.L.; Deng, J.; et al. Discovery of Thieno[3,2-d]Pyrimidine-6-Carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Chemical Computing Group ULC Molecular Operating Environment (MOE2019.01); Chemical Computing Group: Montreal, QC, Canada, 2021. [Google Scholar]
- Liu, F.; Yang, N. Multiscale Landscape of Molecular Mechanism of SIRT1 Activation by STACs. Phys. Chem. Chem. Phys. 2020, 22, 826–837. [Google Scholar] [CrossRef]
- Sharma, A.; Gautam, V.K.; Costantini, S.; Paladino, A.; Colonna, G. Interactomic and Pharmacological Insights on Human Sirt-1. Front. Pharmacol. 2012, 3, 40. [Google Scholar] [CrossRef]
- Manna, D.; Bhuyan, R.; Ghosh, R. Probing the Mechanism of SIRT1 Activation by a 1,4-Dihydropyridine. J. Mol. Model. 2018, 24, 340. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Rooklin, D.; Fang, H.; Zhang, Y. Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation. Sci. Rep. 2016, 6, 38186. [Google Scholar] [CrossRef]
- Liu, F.; Pang, N.; Xu, R.-M.; Yang, N. Mechanism and Design of Allosteric Activators of SIRT1. Protein Cell 2022, 14, 387–392. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 Inhibition by Ligand-Induced Rearrangement of the Active Site. Nat. Commun. 2015, 6, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Knyphausen, P.; de Boor, S.; Kuhlmann, N.; Scislowski, L.; Extra, A.; Baldus, L.; Schacherl, M.; Baumann, U.; Neundorf, I.; Lammers, M. Insights into Lysine Deacetylation of Natively Folded Substrate Proteins by Sirtuins. J. Biol. Chem. 2016, 291, 14677–14694. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the Histone Deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fung, Y.M.E.; Zhang, W.; He, B.; Chung, M.W.H.; Jin, J.; Hu, J.; Lin, H.; Hao, Q. Deacylation Mechanism by SIRT2 Revealed in the 1′-SH-2′-O-Myristoyl Intermediate Structure. Cell Chem. Biol. 2017, 24, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; He, B.; Zhang, X.; Lin, H.; Wang, Y. SIRT2 Reverses 4-Oxononanoyl Lysine Modification on Histones. J. Am. Chem. Soc. 2016, 138, 12304–12307. [Google Scholar] [CrossRef]
- RCSB PDB—6L66: Sirtuin 2 Protein with H3K18 Myristoylated Peptide and Intact NAD Molecule. Available online: https://www.rcsb.org/structure/6L66 (accessed on 31 July 2023).
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD+ Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB—6L65: Sirtuin 2 Protein with H3K18 Myristoylated Peptide. Available online: https://www.rcsb.org/structure/6L65 (accessed on 31 July 2023).
- RCSB PDB—6L71: Sirtuin 2 Demyristoylation Native Intermediate I & II Mixture. Available online: https://www.rcsb.org/structure/6L71 (accessed on 31 July 2023).
- RCSB PDB—6L72: Sirtuin 2 Demyristoylation Native Final Product. Available online: https://www.rcsb.org/structure/6L72 (accessed on 31 July 2023).
- Yamagata, K.; Goto, Y.; Nishimasu, H.; Morimoto, J.; Ishitani, R.; Dohmae, N.; Takeda, N.; Nagai, R.; Komuro, I.; Suga, H.; et al. Structural Basis for Potent Inhibition of SIRT2 Deacetylase by a Macrocyclic Peptide Inducing Dynamic Structural Change. Structure 2014, 22, 345–352. [Google Scholar] [CrossRef]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.E.; Lam, E.W.-F.; Steegborn, C.; et al. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wang, H.-L.; Zhong, L.; Yuan, C.; Liu, S.-Y.; Yu, Z.-J.; Liu, S.; Yan, Y.-H.; Wu, C.; Wang, Y.; et al. X-ray Crystal Structure Guided Discovery of New Selective, Substrate-Mimicking Sirtuin 2 Inhibitors That Exhibit Activities against Non-Small Cell Lung Cancer Cells. Eur. J. Med. Chem. 2018, 155, 806–823. [Google Scholar] [CrossRef]
- Kudo, N.; Ito, A.; Arata, M.; Nakata, A.; Yoshida, M. Identification of a Novel Small Molecule That Inhibits Deacetylase but Not Defatty-Acylase Reaction Catalysed by SIRT2. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170070. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-B.; Jing, H.; Aramsangtienchai, P.; He, B.; Khan, S.; Hu, J.; Lin, H.; Hao, Q. Efficient Demyristoylase Activity of SIRT2 Revealed by Kinetic and Structural Studies. Sci. Rep. 2015, 5, 8529. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Zheng, W.; Weiss, S.; Chua, K.F.; Steegborn, C. Structural Basis for the Activation and Inhibition of Sirtuin 6 by Quercetin and Its Derivatives. Sci. Rep. 2019, 9, 19176. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-Based Development of an Affinity Probe for Sirtuin 2. Angew. Chem. Int. Ed. Engl. 2016, 55, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Price, I.R.; Bai, J.J.; Lin, H. A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol. 2019, 14, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.L.; Rajabi, N.; Kudo, N.; Lundø, K.; Moreno-Yruela, C.; Bæk, M.; Fontenas, M.; Lucidi, A.; Madsen, A.S.; Yoshida, M.; et al. Mechanism-Based Inhibitors of SIRT2: Structure–Activity Relationship, X-Ray Structures, Target Engagement, Regulation of α-Tubulin Acetylation and Inhibition of Breast Cancer Cell Migration. RSC Chem. Biol. 2021, 2, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure–Activity Relationship, X-Ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef] [PubMed]
- Scarano, N.; Abbotto, E.; Musumeci, F.; Salis, A.; Brullo, C.; Fossa, P.; Schenone, S.; Bruzzone, S.; Cichero, E. Virtual Screening Combined with Enzymatic Assays to Guide the Discovery of Novel SIRT2 Inhibitors. Int. J. Mol. Sci. 2023, 24, 9363. [Google Scholar] [CrossRef]
- Huhtiniemi, T.; Suuronen, T.; Rinne, V.M.; Wittekindt, C.; Lahtela-Kakkonen, M.; Jarho, E.; Wallén, E.A.A.; Salminen, A.; Poso, A.; Leppänen, J. Oxadiazole-Carbonylaminothioureas as SIRT1 and SIRT2 Inhibitors. J. Med. Chem. 2008, 51, 4377–4380. [Google Scholar] [CrossRef]
- Huhtiniemi, T.; Wittekindt, C.; Laitinen, T.; Leppänen, J.; Salminen, A.; Poso, A.; Lahtela-Kakkonen, M. Comparative and Pharmacophore Model for Deacetylase SIRT1. J. Comput. Aided Mol. Des. 2006, 20, 589–599. [Google Scholar] [CrossRef]
- Wang, F.; Liu, D.; Wang, H.; Luo, C.; Zheng, M.; Liu, H.; Zhu, W.; Luo, X.; Zhang, J.; Jiang, H. Computational Screening for Active Compounds Targeting Protein Sequences: Methodology and Experimental Validation. J. Chem. Inf. Model. 2011, 51, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Specs—Compound Management Services and Research Compounds for the Life Science Industry. Available online: https://www.specs.net/index.php (accessed on 31 July 2023).
- Alvala, M.; Bhatnagar, S.; Ravi, A.; Jeankumar, V.U.; Manjashetty, T.H.; Yogeeswari, P.; Sriram, D. Novel Acridinedione Derivatives: Design, Synthesis, SIRT1 Enzyme and Tumor Cell Growth Inhibition Studies. Bioorg. Med. Chem. Lett. 2012, 22, 3256–3260. [Google Scholar] [CrossRef] [PubMed]
- Pulla, V.K.; Alvala, M.; Sriram, D.S.; Viswanadha, S.; Sriram, D.; Yogeeswari, P. Structure-Based Drug Design of Small Molecule SIRT1 Modulators to Treat Cancer and Metabolic Disorders. J. Mol. Graph. Model. 2014, 52, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Ramu, M.; Mathur, S.; Unni, S.; Thiyagarajan, S. Identification of New Inhibitors for Human SIRT1: An in-Silico Approach. Med. Chem. 2016, 12, 347–361. [Google Scholar] [CrossRef]
- Wössner, N.; Alhalabi, Z.; González, J.; Swyter, S.; Gan, J.; Schmidtkunz, K.; Zhang, L.; Vaquero, A.; Ovaa, H.; Einsle, O.; et al. Sirtuin 1 Inhibiting Thiocyanates (S1th)—A New Class of Isotype Selective Inhibitors of NAD+ Dependent Lysine Deacetylases. Front. Oncol. 2020, 10, 657. [Google Scholar] [CrossRef]
- Flori, L.; Petrarolo, G.; Brogi, S.; La Motta, C.; Testai, L.; Calderone, V. Identification of Novel SIRT1 Activators Endowed with Cardioprotective Profile. Eur. J. Pharm. Sci. 2021, 165, 105930. [Google Scholar] [CrossRef] [PubMed]
- Manikanttha, M.; Deepti, K.; Tej, M.B.; Reddy, A.G.; Kapavarapu, R.; Rao, M.V.B.; Pal, M. Wang Resin Catalyzed Green Synthesis of 1,8-Dioxo-Octahydroxanthene Derivatives and Their in Silico/in Vitro Evaluation against SIRT1. J. Mol. Struct. 2022, 1264, 133313. [Google Scholar] [CrossRef]
- Purushotham, N.; Singh, M.; Paramesha, B.; Kumar, V.; Wakode, S.; Banerjee, S.K.; Poojary, B.; Asthana, S. Design and Synthesis of Amino Acid Derivatives of Substituted Benzimidazoles and Pyrazoles as Sirt1 Inhibitors. RSC Adv. 2022, 12, 3809–3827. [Google Scholar] [CrossRef]
- Cai, H.; Wang, Y.; Zhang, J.; Wei, Z.; Yan, T.; Feng, C.; Xu, Z.; Zhou, A.; Wu, Y. Discovery of Novel SIRT1/2 Inhibitors with Effective Cytotoxicity against Human Leukemia Cells. J. Chem. Inf. Model. 2023, 63, 4780–4790. [Google Scholar] [CrossRef]
- Pulla, V.K.; Sriram, D.S.; Viswanadha, S.; Sriram, D.; Yogeeswari, P. Energy-Based Pharmacophore and Three-Dimensional Quantitative Structure-Activity Relationship (3D-QSAR) Modeling Combined with Virtual Screening to Identify Novel Small-Molecule Inhibitors of Silent Mating-Type Information Regulation 2 Homologue 1 (SIRT1). J. Chem. Inf. Model. 2016, 56, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Azminah, A.; Erlina, L.; Radji, M.; Mun’im, A.; Syahdi, R.R.; Yanuar, A. In Silico and in Vitro Identification of Candidate SIRT1 Activators from Indonesian Medicinal Plants Compounds Database. Comput. Biol. Chem. 2019, 83, 107096. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Ohue, M.; Gryniukova, A.; Borysko, P.; Zozulya, S.; Yasuo, N.; Yoshino, R.; Ikeda, K.; Shin, W.H.; Kihara, D.; et al. A Prospective Compound Screening Contest Identified Broader Inhibitors for Sirtuin 1. Sci. Rep. 2019, 9, 19585. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int. J. Mol. Sci. 2022, 23, 15105. [Google Scholar] [CrossRef] [PubMed]
- Asinex.Com—Home. Available online: https://www.asinex.com/ (accessed on 13 May 2022).
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Princeton BioMolecular Research, Inc. Available online: https://princetonbio.com/ (accessed on 13 May 2022).
- Mulakayala, N.; Murthy, P.V.N.S.; Rambabu, D.; Aeluri, M.; Adepu, R.; Krishna, G.R.; Reddy, C.M.; Prasad, K.R.S.; Chaitanya, M.; Kumar, C.S.; et al. Catalysis by Molecular Iodine: A Rapid Synthesis of 1,8-Dioxo-Octahydroxanthenes and Their Evaluation as Potential Anticancer Agents. Bioorg. Med. Chem. Lett. 2012, 22, 2186–2191. [Google Scholar] [CrossRef]
- Al-Omran, F.; Mohareb, R.M.; El-Khair, A.A. New Route for Synthesis, Spectroscopy, and X-Ray Studies of 2-[Aryl-(6′-Hydroxy-4′,4′-Dimethyl-2′-Oxocyclohex-6′-Enyl)Methyl]-3-Hydroxy-5,5-Dimethylcyclohex-2-Enone and 1,8-Dioxo-Octahydroxanthenes and Antitumor Evaluation. Med. Chem. Res. 2014, 23, 1623–1633. [Google Scholar] [CrossRef]
- Menezes, A.P.d.J.; Silva, M.L.d.; Pereira, W.L.; Costa, G.d.P.; Horta, A.L.; Mendonça, A.A.S.; Carneiro, A.C.A.; Souza, D.M.S.d.; Novaes, R.D.; Teixeira, R.R.; et al. In Vitro Tripanocidal Effect of 1,8-Dioxooctahydroxanthenes (Xanthenodiones) and Tetraketones and Improvement of Cardiac Parameters in Vivo. J. Glob. Antimicrob. Resist. 2020, 22, 466–476. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, Q.; Zhang, L.; Wang, J.; Shu, M.; Huang, K.; Hu, Y.; Lin, Z. Virtual Screening Identifies Tipranavir as a SIRT1 Inhibitor with Anti-Hepatocarcinoma Effect. Future Med. Chem. 2023, 15, 437–451. [Google Scholar] [CrossRef]
- Basis Data Tanaman Obat Indonesia. Available online: http://herbaldb.farmasi.ui.ac.id/v3/ (accessed on 13 May 2022).
- Tervo, A.J.; Kyrylenko, S.; Niskanen, P.; Salminen, A.; Leppänen, J.; Nyrönen, T.H.; Järvinen, T.; Poso, A. An In Silico Approach to Discovering Novel Inhibitors of Human Sirtuin Type 2. J. Med. Chem. 2004, 47, 6292–6298. [Google Scholar] [CrossRef]
- Maybridge Library|Thermo Fisher Scientific—IT. Available online: https://www.thermofisher.com/it/en/home/global/forms/lab-solutions/maybridge-library.html (accessed on 13 May 2022).
- Tervo, A.J.; Suuronen, T.; Kyrylenko, S.; Kuusisto, E.; Kiviranta, P.H.; Salminen, A.; Leppanen, J.; Poso, A. Discovering Inhibitors of Human Sirtuin Type 2: Novel Structural Scaffolds. J. Med. Chem. 2006, 49, 7239–7241. [Google Scholar] [CrossRef]
- Sivaraman, P.; Mattegunta, S.; Subbaraju, G.V.; Satyanarayana, C.; Padmanabhan, B. Design of a Novel Nucleoside Analog as Potent Inhibitor of the NAD+ Dependent Deacetylase, SIRT2. Syst. Synth. Biol. 2010, 4, 257–263. [Google Scholar] [CrossRef]
- Schlicker, C.; Boanca, G.; Lakshminarasimhan, M.; Steegborn, C. Structure-Based Development of Novel Sirtuin Inhibitors. Aging 2011, 3, 852–872. [Google Scholar] [CrossRef]
- Sacconnay, L.; Ryckewaert, L.; Randazzo, G.M.; Petit, C.; Passos, C.D.S.; Jachno, J.; Michailovienė, V.; Zubrienė, A.; Matulis, D.; Carrupt, P.-A.; et al. 5-Benzylidene-Hydantoin Is a New Scaffold for SIRT Inhibition: From Virtual Screening to Activity Assays. Eur. J. Pharm. Sci. 2016, 85, 59–67. [Google Scholar] [CrossRef]
- Abbotto, E.; Casini, B.; Piacente, F.; Scarano, N.; Cerri, E.; Tonelli, M.; Astigiano, C.; Millo, E.; Sturla, L.; Bruzzone, S.; et al. Novel Thiazole-Based SIRT2 Inhibitors Discovered via Molecular Modelling Studies and Enzymatic Assays. Pharmaceuticals 2023, 16, 1316. [Google Scholar] [CrossRef]
- Uciechowska, U.; Schemies, J.; Neugebauer, R.C.; Huda, E.-M.; Schmitt, M.L.; Meier, R.; Verdin, E.; Jung, M.; Sippl, W. Thiobarbiturates as Sirtuin Inhibitors: Virtual Screening, Free-Energy Calculations, and Biological Testing. ChemMedChem 2008, 3, 1965–1976. [Google Scholar] [CrossRef]
- Neugebauer, R.C.; Uchiechowska, U.; Meier, R.; Hruby, H.; Valkov, V.; Verdin, E.; Sippl, W.; Jung, M. Structure–Activity Studies on Splitomicin Derivatives as Sirtuin Inhibitors and Computational Prediction of Binding Mode. J. Med. Chem. 2008, 51, 1203–1213. [Google Scholar] [CrossRef]
- Eren, G.; Bruno, A.; Guntekin-Ergun, S.; Cetin-Atalay, R.; Ozgencil, F.; Ozkan, Y.; Gozelle, M.; Kaya, S.G.; Costantino, G. Pharmacophore Modeling and Virtual Screening Studies to Identify Novel Selective SIRT2 Inhibitors. J. Mol. Graph. Model. 2019, 89, 60–73. [Google Scholar] [CrossRef]
- Khanfar, M.A.; Alqtaishat, S. Discovery of Potent Natural-Product-Derived SIRT2 Inhibitors Using Structure- Based Exploration of SIRT2 Pharmacophoric Space Coupled With QSAR Analyses. Anticancer. Agents Med. Chem. 2021, 21, 2278–2286. [Google Scholar] [CrossRef]
- Djokovic, N.; Ruzic, D.; Rahnasto-Rilla, M.; Srdic-Rajic, T.; Lahtela-Kakkonen, M.; Nikolic, K. Expanding the Accessible Chemical Space of SIRT2 Inhibitors through Exploration of Binding Pocket Dynamics. J. Chem. Inf. Model. 2022, 62, 2571–2585. [Google Scholar] [CrossRef]
- Kiviranta, P.H.; Leppänen, J.; Rinne, V.M.; Suuronen, T.; Kyrylenko, O.; Kyrylenko, S.; Kuusisto, E.; Tervo, A.J.; Järvinen, T.; Salminen, A.; et al. N-(3-(4-Hydroxyphenyl)-Propenoyl)-Amino Acid Tryptamides as SIRT2 Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2448–2451. [Google Scholar] [CrossRef]
- Manjula, R.; Gokhale, N.; Unni, S.; Deshmukh, P.; Reddyrajula, R.; Srinivas Bharath, M.M.; Dalimba, U.; Padmanabhan, B. Design, Synthesis, in-Vitro Evaluation and Molecular Docking Studies of Novel Indole Derivatives as Inhibitors of SIRT1 and SIRT2. Bioorg. Chem. 2019, 92, 103281. [Google Scholar] [CrossRef]
- Home—NCI Wiki. Available online: https://wiki.nci.nih.gov/dashboard.action (accessed on 13 May 2022).
- Schuetz, A.; Min, J.; Antoshenko, T.; Wang, C.L.; Allali-Hassani, A.; Dong, A.; Loppnau, P.; Vedadi, M.; Bochkarev, A.; Sternglanz, R.; et al. Structural Basis of Inhibition of the Human NAD+-Dependent Deacetylase SIRT5 by Suramin. Structure 2007, 15, 377–389. [Google Scholar] [CrossRef]
- Pan, P.W.; Feldman, J.L.; Devries, M.K.; Dong, A.; Edwards, A.M.; Denu, J.M. Structure and Biochemical Functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. [Google Scholar] [CrossRef]
- ChemBridge|Home. Available online: https://www.chembridge.com/ (accessed on 13 May 2022).
- Uciechowska, U.; Schemies, J.; Scharfe, M.; Lawson, M.; Wichapong, K.; Jung, M.; Sippl, W. Binding Free Energy Calculations and Biological Testing of Novel Thiobarbiturates as Inhibitors of the Human NAD+ Dependent Histone Deacetylase Sirt2. Medchemcomm 2012, 3, 167–173. [Google Scholar] [CrossRef]
- Yagci, S.; Gozelle, M.; Kaya, S.G.; Ozkan, Y.; Aksel, A.B.; Bakar-Ates, F.; Dundar, Y.; Eren, G. Hit-to-Lead Optimization on Aryloxybenzamide Derivative Virtual Screening Hit against SIRT. Bioorg. Med. Chem. 2021, 30, 115961. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Rogers, D.; Hopfinger, A.J. Application of Genetic Function Approximation to Quantitative Structure-Activity Relationships and Quantitative Structure-Property Relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Taha, M.O.; Bustanji, Y.; Al-Ghussein, M.A.S.; Mohammad, M.; Zalloum, H.; Al-Masri, I.M.; Atallah, N. Pharmacophore Modeling, Quantitative Structure–Activity Relationship Analysis, and in Silico Screening Reveal Potent Glycogen Synthase Kinase-3β Inhibitory Activities for Cimetidine, Hydroxychloroquine, and Gemifloxacin. J. Med. Chem. 2008, 51, 2062–2077. [Google Scholar] [CrossRef]
- MEGx Purified Natural Product Screening Compounds—AnalytiCon Discovery. Available online: https://ac-discovery.com/purified-natural-product-screening-compounds/ (accessed on 13 May 2022).
- Karaman, B.; Alhalabi, Z.; Swyter, S.; Mihigo, S.; Andrae-Marobela, K.; Jung, M.; Sippl, W.; Ntie-Kang, F. Identification of Bichalcones as Sirtuin Inhibitors by Virtual Screening and In Vitro Testing. Molecules 2018, 23, 416. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Amoa Onguéné, P.; Fotso, G.W.; Andrae-Marobela, K.; Bezabih, M.; Ndom, J.C.; Ngadjui, B.T.; Ogundaini, A.O.; Abegaz, B.M.; Meva’a, L.M. Virtualizing the P-ANAPL Library: A Step towards Drug Discovery from African Medicinal Plants. PLoS ONE 2014, 9, e90655. [Google Scholar] [CrossRef]
- Wang, J.; Zang, W.; Liu, J.; Zheng, W. Bivalent SIRT1 Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 180–186. [Google Scholar] [CrossRef]
- Laxmi, D.S.; Vardhini, S.V.; Guttikonda, V.R.; Rao, M.V.B.; Pal, M. Synthesis of 2-Substituted Furo [3,2-b]Pyridines Under Pd/C-Cu Catalysis Assisted by Ultrasound: Their Evaluation as Potential Cytotoxic Agents. Anticancer. Agents Med. Chem. 2020, 20, 932–940. [Google Scholar] [CrossRef]
- Laaroussi, H.; Ding, Y.; Teng, Y.; Deschamps, P.; Vidal, M.; Yu, P.; Broussy, S. Synthesis of Indole Inhibitors of Silent Information Regulator 1 (SIRT1), and Their Evaluation as Cytotoxic Agents. Eur. J. Med. Chem. 2020, 202, 112561. [Google Scholar] [CrossRef]
- Li, C.; Hu, S.-S.; Yang, L.; Wang, M.; Long, J.-D.; Wang, B.; Han, H.; Zhu, H.; Zhao, S.; Liu, J.-G.; et al. Discovery of 5-Benzylidene-2-Phenyl-1,3-Dioxane-4,6-Diones as Highly Potent and Selective SIRT1 Inhibitors. ACS Med. Chem. Lett. 2021, 12, 397–403. [Google Scholar] [CrossRef]
- Challa, C.S.; Katari, N.K.; Nallanchakravarthula, V.; Nayakanti, D.; Kapavarapu, R.; Pal, M. Amberlyst-15 Catalysed Sonochemical Synthesis of 2-Amino-4,6-Disubstituted Nicotinonitrile Derivatives and Their Biological Evaluation. J. Mol. Struct. 2021, 1240, 130541. [Google Scholar] [CrossRef]
- Challa, C.S.; Katari, N.K.; Nallanchakravarthula, V.; Nayakanti, D.; Kapavarapu, R.; Pal, M. Wang-OSO3H Catalyzed Green Synthesis of 2-Arylamino-3-Cyanopyridine Derivatives under Ultrasound: Their Assessment as Potential Inhibitors of SIRT1. J. Mol. Struct. 2022, 1253, 132309. [Google Scholar] [CrossRef]
- Kondabanthini, S.; Akshinthala, P.; Katari, N.K.; Srimannarayana, M.; Gundla, R.; Kapavarapu, R.; Pal, M. A Rapid Synthesis of 5-Substituted 7-Amino-6-Cyano-1,5-Dihydro-1H-Pyrano [2,3-d]Pyrimidine-2,4(3H)-Diones and Their in Silico/Evaluation against SIRT1. J. Mol. Struct. 2023, 1276, 134753. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, H.; Yu, X.; Peng, T.; Wang, G.; Wen, X.; Sun, Y.; Liu, S.; Zhang, S.; Hu, L.; et al. Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors. Molecules 2017, 22, 1348. [Google Scholar] [CrossRef]
- Seifert, T.; Malo, M.; Kokkola, T.; Stéen, E.J.L.; Meinander, K.; Wallén, E.A.A.; Jarho, E.M.; Luthman, K. A Scaffold Replacement Approach towards New Sirtuin 2 Inhibitors. Bioorg. Med. Chem. 2020, 28, 115231. [Google Scholar] [CrossRef]
- Chowdhury, S.; Sripathy, S.; Webster, A.; Park, A.; Lao, U.; Hsu, J.H.; Loe, T.; Bedalov, A.; Simon, J.A. Discovery of Selective SIRT2 Inhibitors as Therapeutic Agents in B-Cell Lymphoma and Other Malignancies. Molecules 2020, 25, 455. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, A.H.; Meng, X.-G.; Marzouk, A.A.; Fouad, A.; Abdelazeem, A.H.; Youssif, B.G.M.; Jiang, H.; Wang, M.-Q. Structure-Based Design, Synthesis, and Biological Evaluation of Novel Piperine–Resveratrol Hybrids as Antiproliferative Agents Targeting SIRT-2. RSC Adv. 2021, 11, 25738–25751. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.; Wilson, D.J.; Chen, L. 5-((3-Amidobenzyl)Oxy)Nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs. Molecules 2023, 28, 7655. [Google Scholar] [CrossRef] [PubMed]
- Mazur, G.; Pańczyk-Straszak, K.; Krysińska, K.; Niemiec, K.; Waszkielewicz, A. Novel Xanthone Derivatives as Potent Sirtuin 2 Inhibitors. Bioorg. Med. Chem. Lett. 2024, 100, 129620. [Google Scholar] [CrossRef]
- Badran, M.M.; Abbas, S.H.; Tateishi, H.; Maemoto, Y.; Toma, T.; Ito, A.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M.; Radwan, M.O. Ligand-Based Design and Synthesis of New Trityl Histamine and Trityl Cysteamine Derivatives as SIRT2 Inhibitors for Cancer Therapy. Eur. J. Med. Chem. 2024, 269, 116302. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.A.; Ahmed, E.M.; Zaher, A.F.; El-Zoghbi, M.S.; Sobh, E.A. Synthesis of Certain Benzothieno [3,2-d]Pyrimidine Derivatives as a Selective SIRT2 Inhibitors. Eur. J. Med. Chem. 2020, 187, 111926. [Google Scholar] [CrossRef]
- Han, H.; Li, C.; Li, M.; Yang, L.; Zhao, S.; Wang, Z.; Liu, H.; Liu, D. Design, Synthesis, and Biological Evaluation of 8-Mercapto-3,7-Dihydro-1H-Purine-2,6-Diones as Potent Inhibitors of SIRT1, SIRT2, SIRT3, and SIRT5. Molecules 2020, 25, 2755. [Google Scholar] [CrossRef]
- Wang, M.; Rakesh, K.P.; Leng, J.; Fang, W.-Y.; Ravindar, L.; Channe Gowda, D.; Qin, H.-L. Amino Acids/Peptides Conjugated Heterocycles: A Tool for the Recent Development of Novel Therapeutic Agents. Bioorg. Chem. 2018, 76, 113–129. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Carafa, V.; Lara, E.; Meade, S.; Botta, G.; Nebbioso, A.; Schemies, J.; Jung, M.; Kazantsev, A.G.; et al. Identification of Tri- and Tetracyclic Pyrimidinediones as Sirtuin Inhibitors. ChemMedChem 2010, 5, 674–677. [Google Scholar] [CrossRef]
- Kondabanthini, S.; Katari, N.K.; Srimannarayana, M.; Gundla, R.; Kapavarapu, R.; Pal, M. Wang Resin Catalyzed Sonochemical Synthesis of Dihydropyrano [2,3-c]Pyrazole Derivatives and Their Interactions with SIRT1. J. Mol. Struct. 2022, 1266, 133527. [Google Scholar] [CrossRef]
- Seifert, T.; Malo, M.; Kokkola, T.; Engen, K.; Fridén-Saxin, M.; Wallén, E.A.A.; Lahtela-Kakkonen, M.; Jarho, E.M.; Luthman, K. Chroman-4-One- and Chromone-Based Sirtuin 2 Inhibitors with Antiproliferative Properties in Cancer Cells. J. Med. Chem. 2014, 57, 9870–9888. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of Potent and Selective Sirtuin 2 (SIRT2) Inhibitors Using a Fragment-Based Approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.; Wilson, D.J.; More, S.S.; Xie, J.; Chen, L. 5-((3-Amidobenzyl)Oxy)Nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 2016, 59, 2928–2941. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and Initial Evaluation of YM-08, a Blood-Brain Barrier Permeable Derivative of the Heat Shock Protein 70 (Hsp70) Inhibitor MKT-077, Which Reduces Tau Levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Yeong, K.Y.; Khaw, K.Y.; Takahashi, Y.; Itoh, Y.; Murugaiyah, V.; Suzuki, T. Discovery of Gamma-Mangostin from Garcinia Mangostana as a Potent and Selective Natural SIRT2 Inhibitor. Bioorg. Chem. 2020, 94, 103403. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.F.S.; Ciftci, H.I.; Radwan, M.O.; Koga, R.; Ohsugi, T.; Okiyama, Y.; Honma, T.; Nakata, A.; Ito, A.; Yoshida, M.; et al. New SIRT2 Inhibitors: Histidine-Based Bleomycin Spin-Off. Bioorg. Med. Chem. 2019, 27, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Di Fruscia, P.; Zacharioudakis, E.; Liu, C.; Moniot, S.; Laohasinnarong, S.; Khongkow, M.; Harrison, I.F.; Koltsida, K.; Reynolds, C.R.; Schmidtkunz, K.; et al. The Discovery of a Highly Selective 5,6,7,8-Tetrahydrobenzo [4,5]Thieno [2,3-d]Pyrimidin-4(3H)-One SIRT2 Inhibitor That Is Neuroprotective in an in Vitro Parkinson’s Disease Model. ChemMedChem 2015, 10, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of Manganese Superoxide Dismutase by Nuclear Translocation and Activation of SIRT1 Promotes Cell Survival in Chronic Heart Failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an Activator of SIRT1, Upregulates Sarcoplasmic Calcium ATPase and Improves Cardiac Function in Diabetic Cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef]
- Kim, H.-D.; Hesterman, J.; Call, T.; Magazu, S.; Keeley, E.; Armenta, K.; Kronman, H.; Neve, R.L.; Nestler, E.J.; Ferguson, D. SIRT1 Mediates Depression-Like Behaviors in the Nucleus Accumbens. J. Neurosci. 2016, 36, 8441–8452. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, F.; Guan, X. Baicalin Reverse Depressive-like Behaviors through Regulation SIRT1-NF-kB Signaling Pathway in Olfactory Bulbectomized Rats. Phytother. Res. 2019, 33, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and Their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef] [PubMed]
- Karaman Mayack, B.; Sippl, W.; Ntie-Kang, F. Natural Products as Modulators of Sirtuins. Molecules 2020, 25, 3287. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing Ageing by Design: The Rise of NAD+ and Sirtuin-Activating Compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin Activators and Inhibitors: Promises, Achievements, and Challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Bursch, K.L.; Goetz, C.J.; Smith, B.C. Current Trends in Sirtuin Activator and Inhibitor Development. Molecules 2024, 29, 1185. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Peptide-based drug discovery: Current status and recent advances. Drug Discov. Today 2023, 28, 2. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Schiedel, M.; Lehotzky, A.; Szunyogh, S.; Oláh, J.; Hammelmann, S.; Wössner, N.; Robaa, D.; Einsle, O.; Sippl, W.; Ovádi, J.; et al. HaloTag-Targeted Sirtuin-Rearranging Ligand (SirReal) for the Development of Proteolysis-Targeting Chimeras (PROTACs) against the Lysine Deacetylase Sirtuin 2 (Sirt2). Chembiochem 2020, 21, 3371–3376. [Google Scholar] [CrossRef]
- Sakkiah, S.; Chandrasekaran, M.; Lee, Y.; Kim, S.; Lee, K.W. Molecular modeling study for conformational changes of Sirtuin 2 due to substrate and inhibitor binding. J. Biomol. Struct. Dyn. 2012, 30, 235–254. [Google Scholar] [CrossRef]
- Li, J.; Flick, F.; Verheugd, P.; Carloni, P.; Lüscher, B.; Rossetti, G. Insight into the Mechanism of Intramolecular Inhibition of the Catalytic Activity of Sirtuin 2 (SIRT2). PLoS ONE 2015, 10, e0139095. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhou, Y.; Wang, S.; Zhang, Y. Sirtuin Deacetylation Mechanism and Catalytic Role of the Dynamic Cofactor Binding Loop. J. Phys. Chem. Lett. 2013, 4, 491–495. [Google Scholar] [CrossRef] [PubMed]
- von der Esch, B.; Dietschreit, J.C.B.; Peters, L.D.M.; Ochsenfeld, C. Finding Reactive Configurations: A Machine Learning Approach for Estimating Energy Barriers Applied to Sirtuin 5. J. Chem. Theory Comput. 2019, 15, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Srivastava, M.; Wakode, S.R.; Asthana, S. Elucidation of Structural Determinants Delineates the Residues Playing Key Roles in Differential Dynamics and Selective Inhibition of Sirt1-3. J. Chem. Inf. Model. 2021, 61, 1105–1124. [Google Scholar] [CrossRef]
- Djokovic, N.; Rahnasto-Rilla, M.; Lougiakis, N.; Lahtela-Kakkonen, M.; Nikolic, K. SIRT2i_Predictor: A Machine Learning-Based Tool to Facilitate the Discovery of Novel SIRT2 Inhibitors. Pharmaceuticals 2023, 16, 127. [Google Scholar] [CrossRef]













| PDB ID | Structure Title | Small Molecule-like Ligand (Selectivity) | Cofactor | Substrate | R (Å) | R.D. | Ref. | L | Additional Domains/ Regions |
|---|---|---|---|---|---|---|---|---|---|
| 4IG9 | Structure of NAD+-dependent protein deacetylase sirtuin-1 (open state) | None | None | None | 2.64 | 2013 | [27] | 281 | C-terminal regulatory segment (CTR) |
| 4KXQ | Structure of NAD+-dependent protein deacetylase sirtuin-1 (closed state) | None | ADPR | None | 1.849 | 2013 | [27] | 281 | C-terminal regulatory segment |
| 4IF6 | Structure of NAD+-dependent protein deacetylase sirtuin-1 (closed state) | None | ADPR | None | 2.25 | 2013 | [35] | 281 | C-terminal regulatory segment (CTR) |
| 5BTR | Crystal structure of SIRT1 in complex with resveratrol and an AMC-containing peptide | Resveratrol (activator) | None | p53-AMC peptide | 3.2 | 2015 | [34] | 397 | N-terminal domain |
| 4I5I | Crystal structure of the SIRT1 catalytic domain bound to NAD+ and an EX527 analog | EX-527 analog (inhibitor)(PS) †,  | NAD+ | None | 2.5 | 2013 | [36] | 287 | |
| 4ZZI | SIRT1/Activator/Inhibitor Complex | (4TQ) activator (1NS) inhibitor (NS ††)  | None | None | 2.73 | 2015 | [37] | 356 | N-terminal domain, CTR |
| 4ZZH | SIRT1/Activator Complex | Activator | None | None | 3.10 | 2015 | [37] | 356 | N-terminal domain + C-terminal regulatory segment (CTR) |
| 4ZZJ | SIRT1/Activator/Substrate Complex | Activator  | Carba-NAD+ | Ac-p53 | 2.74 | 2015 | [37] | 356 | N-terminal domain |
| PDB ID | Cofactor | Substrate (Intermediate/Reaction Product) | R (Å) | R.D. | Ref. | L | PDB ID | Cofactor | Substrate (Intermediate/Reaction Product) | R (Å) | R.D. | Ref. | L |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1J8F | None | None | 1.70 | 2001 | [47] | 323 | 4X3O | None | 1′-SH-2′-O-myristoyl ADPR (covalent intermediate) | 1.50 | 2016 | [48] | 304 |
| 3ZGO | None | None | 1.63 | 2013 | [28] | 325 | 5FYQ | None | 13-mer trifluoroacetylated Ran peptide (modified substrate) | 3.00 | 2016 | [46] | 360 |
| 3ZGV | ADPR | None | 2.27 | 2013 | [28] | 325 | 5G4C | Carba-NAD+ | 4-oxononanoyl peptide | 2.10 | 2017 | [49] | 323 |
| 5D7O | ADPR | None | 1.63 | 2015 | [26] | 310 | 6L66 | NAD+ | H3K18 myristoylated peptide | 2.17 | 2020 | [50] | 304 |
| 4Y6Q | None | 2′-O-myristoyl-ADP-ribose (reaction product), None | 1.90 | 2016 | [51] | 293 | 6L65 | None | H3K18 myristoylated peptide | 1.80 | 2020 | [52] | 307 |
| 4Y6L | None | H3K9myr peptide | 1.60 | 2016 | [51] | 293 | 6L71 | None | Reaction intermediate I and II (c.ADPR and NAM) | 2.11 | 2021 | [53] | 304 |
| 4Y6O | None | Myristoylated peptide (TNF-alphaK20myr) | 1.60 | 2016 | [51] | 293 | 6L72 | None | Final product (NAD+-condensed Ac-R) | 2.50 | 2021 | [54] | 304 |
| PDB ID (R) | Inhibitor (Selectivity) | Cofactor/ Substrate | R.D. | Ref. | L | PDB ID | Inhibitor (Selectivity) | Cofactor/ Substrate | R.D. | Ref. | L |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4L3O (R = 2.52) | Macrocyclic peptide S2iL5 (inhibitor) (NS) | None | 2013 | [55] | 302 | 5MAT (R = 2.07) | Thienopyrimidinone-inhibitor (S)  | None | 2017 | [56] | 303 |
| 4RMG (R = 1.88) | (SirReal2) (S)  | NAD+ | 2015 | [45] | 304 | 5Y5N (R = 2.30) | Anilinobenzamide derivative(S)  | None | 2017 | [20] | 336 |
| 4RMJ (R = 1.87) | Nicotinamide | ADPR | 2015 | [45] | 304 | 5YQM (R = 1.74) | A29-inhibitor (S)  | None | 2018 | [57] | 306 |
| 5D7P (R = 1.76) | EX-243 (NS) | ADPR | 2015 | [26] | 304 | 5YQO (R = 1.48) | L5C-inhibitor (S) | None | 2018 | [57] | 306 |
| 4RMI (R = 1.45) | (SirReal1) (S) | None Ac-Lys-OTC peptide | 2015 | [45] | 304 | 5YQL (R = 1.60) | A2I-inhibitor (S) | None | 2018 | [57] | 306 |
| 4RMH (R = 1.42) | (SirReal2) (S) | None Ac-Lys-H3 peptide | 2015 | [45] | 304 | 5YQN (R = 1.60) | L55-inhibitor (S) | None | 2018 | [57] | 306 |
| 5D7Q (R = 2.01) | inhibitor, (CHIC35) (PS) | ADPR | 2015 | [26] | 304 | 5Y0Z (R = 2.00) | NPD11033-inhibitor (S)  | None | 2018 | [58] | 293 |
| 4R8M (R = 2.10) | BHJH-TM1 (thiomyristoyl peptide-mechanism-based inhibitor) | None | 2015 | [59] | 319 | 6QCN (R = 2.23) | Quercetin | ADPR | 2019 | [60] | 304 |
| 5DY5 (R = 1.95) | Sirt2-IN-1 (S)  | None | 2016 | [61] | 304 | 6NR0 (R = 2.45) | Glucose-TM-1beta glucose-conjugated Thiomyristoyl lysine (mechanism-based inhibitor), covalently bound to cofactor | ADPR (fused to glucose TM-1beta) | 2020 | [62] | 319 |
| 5DY4 (R = 1.77) | SirReal2 analog (S) | NAD+ | 2016 | [63] | 304 | 7BOS (R = 1.7) | Myristoyl thiourea inhibitor n. 13 (mechanism-based inhibitor) | None | 2021 | [64] | 293 |
| 4X3P (R = 1.80) | Peptide-like inhibitor (BHJH-TM1) | Carba-NAD+ | 2016 | [48] | 304 | 7BOT (R = 1.7) | Myristoyl thiourea inhibitor, No.23 (mechanism-based inhibitor) | None | 2021 | [64] | 293 |
| 5MAR (R = 1.89) | (Oxadiazole inhibitor) (S) | ADPR | 2017 | [65] | 303 |
| Entry | R.D. | Ref. | Type of Computational Method | Selectivity over Other Isoforms | Screened Database (n. of Compounds) | Most Active Compound/Proposed Compound | Activator/Inhibitor | Potency |
|---|---|---|---|---|---|---|---|---|
| 1 | 2004 | [91] | Queries: (SB) features calculated on MD generated conformation + Docking | Not tested | Maybridge library |  | I | 56.7 μM (IC50) |
| 2 | 2006 | [93] | Queries: (SB) features calculated on MD generated conformation + Docking | Not tested | Maybridge Screening Collection and LeadQuest libraries |  Indole derivative | I | 51 μM (IC50) |
| 3 | 2010 | [94] | SBVS | 1 | NCI Diversity Set II |  Nucleoside analog (thieno[2,3-d]pyrimidine) | I | 8.7 μM (IC50) |
| 4 | 2011 | [95] | Multi-target SBVS | 3,5,6 | NCI diversity set (1990) |  CSC8 | I | 4.8 μM (SIRT2 IC50) |
| 5 | 2016 | [96] | SBVS | 1 | SPECS library (197,477) |  5-benzylidene-hydantoin | I | 37.7 μM (IC50) |
| 6 | 2023 | [66] | SBVS | Not tested | ChemDIV (22,000) |  L407-0319 | I | 44.3% inhibition at 150 μM |
| 7 | 2023 | [97] | SBVS | 6 | In-house library | 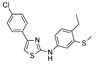 | I | 17.3 μM (IC50) |
| 8 | 2008 | [98] | LB: similarity based | 1 | Chembridge database (~328, 000) |  Thiobarbiturates | I | 9.1 μM (IC50) |
| 9 | 2008 | [99] | LB: similarity based | Not tested | Chembridge (~328,000) |  Lactame analogs of beta-phenylsplitomicins | I | 6.4 μM (IC50) |
| 10 | 2019 | [100] | LB: pharmacophore based | 1,3,5 | ZINC drug-like database (13,000,000) |  ZINC05417772 | I | 84.28% inhibition at 300 μM |
| 11 | 2021 | [101] | SBVS pharmacophore model combined with (SB+LB) QSAR | Not tested | AnalytiCon Discovery database of purified natural products (5637) | 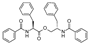 Asperphenamate | I | 1.94 μM (IC50) |
| 12 | 2022 | [102] | SBVS on a linear model combining X-rays, MD-derived conformations, and MetaD-derived conformations | 1,6 | SPECS (200,000) |  NDJ18 | I | 58.7 μM (IC50) |
| Entry | R.D. | Ref. | SIRT(s) | Type of Computational Method | Selectivity over Other Isoforms | Screened Database (n. of Compounds) | Most Active Compound/Proposed Compound | Activator/Inhibitor | Potency |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2018 | [115] | 1,2 | SBVS on different conformations of sirt-1 and sirt-2 | 3 | Pan-African Natural Products Library (463) |  | I | 40.8 μM (SIRT1 IC50) 44.8 μM (SIRT2 IC50,) |
| Type of Inhibitors | Chemical Scaffold | References |
|---|---|---|
| SIRT1Is | Tripeptide linked to heterocyclic nuclei | [117] |
| Furopyridine derivatives | [118] | |
| Indole compounds (strictly related to EX-527) | [119] | |
| Benzylidene–dioxane compounds | [120] | |
| Pyridine derivatives | [121,122] | |
| Tryptophan conjugates | [77] | |
| Pyrano[2,3- d ]pyrimidinone derivatives | [123] | |
| Variable five-membered ring derivatives | [81] | |
| Thienopyrimidone derivatives | [74] | |
| 1,8-dioxo-octahydroxanthene derivatives | [76] | |
| SIRT2Is | Pyrimidine derivatives | [95] |
| 1,2,4-oxadiazole substituted analogs | [65] | |
| Benzofuran derivatives | [124] | |
| Thienopyrimidinone compounds | [56] | |
| Benzothiadiazine-1,1-dioxide-based compounds | [125] | |
| Cambinol-related compounds | [126] | |
| Piperine–resveratrol compounds | [127] | |
| Nicotinamide-containing compounds | [128] | |
| Pyrazolo–pyrimidine derivatives | [66] | |
| Thiazole-based compounds | [97] | |
| Xanthone derivatives | [129] | |
| Cysteamine derivatives | [130] | |
| Dual SIRT1/2Is | Indole and triazole derivatives, | [104] |
| Benzothieno[3,2-d]pyrimidines, | [131] | |
| Naphthyl-based compounds | [78] | |
| Pan-SIRTIs | 8-mercapto-3,7-dihydro-1H-purine-2,6-dione derivatives | [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarano, N.; Brullo, C.; Musumeci, F.; Millo, E.; Bruzzone, S.; Schenone, S.; Cichero, E. Recent Advances in the Discovery of SIRT1/2 Inhibitors via Computational Methods: A Perspective. Pharmaceuticals 2024, 17, 601. https://doi.org/10.3390/ph17050601
Scarano N, Brullo C, Musumeci F, Millo E, Bruzzone S, Schenone S, Cichero E. Recent Advances in the Discovery of SIRT1/2 Inhibitors via Computational Methods: A Perspective. Pharmaceuticals. 2024; 17(5):601. https://doi.org/10.3390/ph17050601
Chicago/Turabian StyleScarano, Naomi, Chiara Brullo, Francesca Musumeci, Enrico Millo, Santina Bruzzone, Silvia Schenone, and Elena Cichero. 2024. "Recent Advances in the Discovery of SIRT1/2 Inhibitors via Computational Methods: A Perspective" Pharmaceuticals 17, no. 5: 601. https://doi.org/10.3390/ph17050601