
Effect of the 35 nm and 70 nm Size Exclusion Chromatography (SEC) Column and Plasma Storage Time on Separated Extracellular Vesicles
 , , , ,
, , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Preparation of PFP from Whole Blood
2.3. Pre-Analytical Measurements of PFP
2.4. Isolation of sEVs
2.5. NTA
2.6. Spectrophotometry
2.7. WB
2.8. TEM
2.9. MS
2.10. Statistical Analysis
2.11. EV-TRACK
3. Results
3.1. Pre-Analytical Measurements of PFP
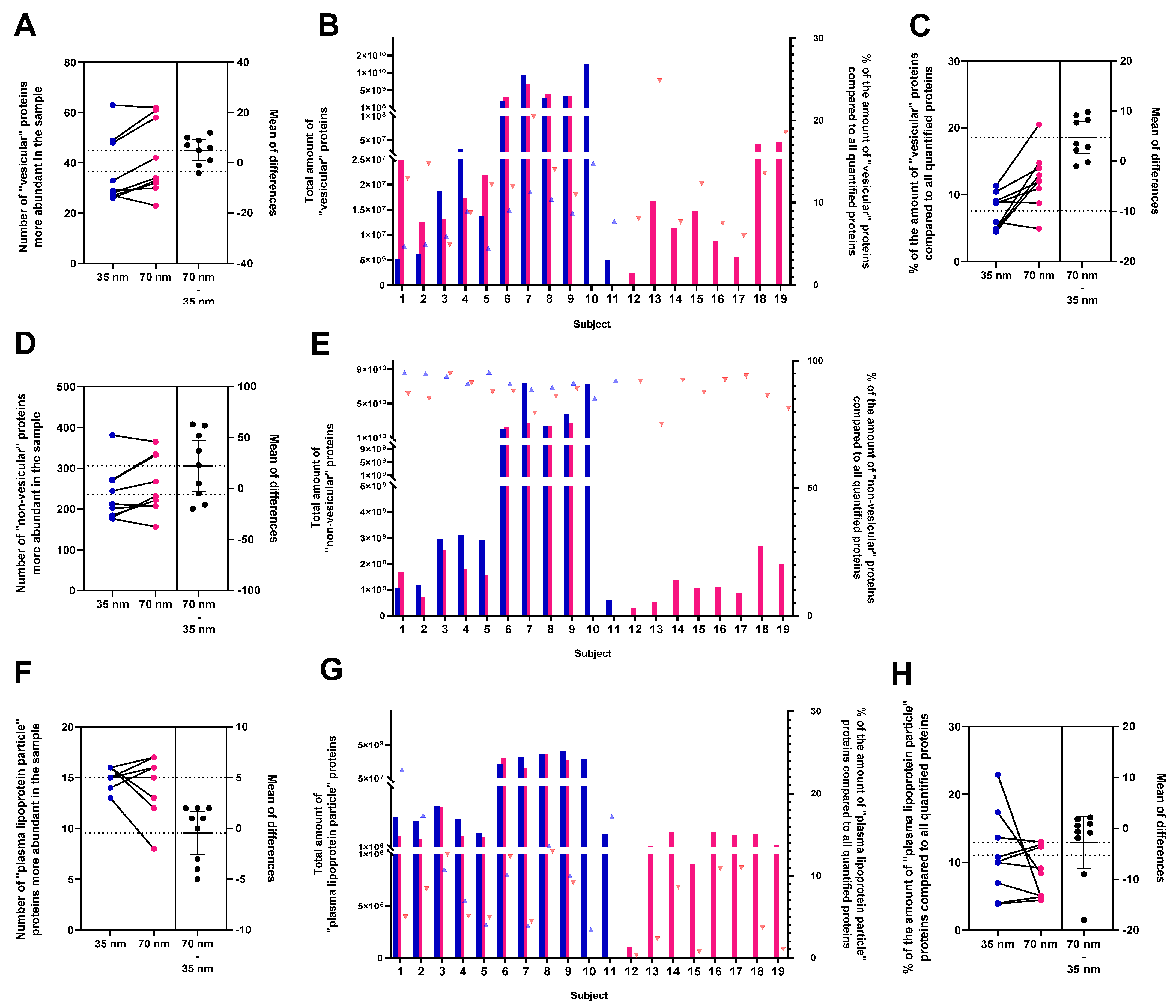
3.2. Characterisation of sEVs Isolated Using SEC on a 35 nm or on a 70 nm Column
3.3. Characterisation of sEVs Separated by an SEC 70 nm Column Both for Fresh and Frozen PFP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Tóth, E.Á.; Turiák, L.; Visnovitz, T.; Cserép, C.; Mázló, A.; Sódar, B.W.; Försönits, A.I.; Petővári, G.; Sebestyén, A.; Komlósi, Z.; et al. Formation of a protein corona on the surface of extracellular vesicles in blood plasma. J. Extracell. Vesicles 2021, 10, e12140. [Google Scholar] [CrossRef]
- Bhutta, M.S.; McInerny, C.J.; Gould, G.W. ESCRT function in cytokinesis: Location, dynamics and regulation by mitotic kinases. Int. J. Mol. Sci. 2014, 15, 21723–21739. [Google Scholar] [CrossRef]
- Olmos, Y. The ESCRT Machinery: Remodeling, Repairing, and Sealing Membranes. Membranes 2022, 12, 633. [Google Scholar] [CrossRef]
- Hurley, J.H.; Emr, S.D. The ESCRT complexes: Structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 277–298. [Google Scholar] [CrossRef]
- Williams, R.L.; Urbé, S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 2007, 8, 355–368. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2022, 23, 236–250. [Google Scholar] [CrossRef]
- Busatto, S.; Morad, G.; Guo, P.; Moses, M.A. The role of extracellular vesicles in the physiological and pathological regulation of the blood-brain barrier. FASEB Bioadv. 2021, 3, 665–675. [Google Scholar] [CrossRef]
- Xu, R.; Fitts, A.; Li, X.; Fernandes, J.; Pochampally, R.; Mao, J.; Liu, Y.M. Quantification of Small Extracellular Vesicles by Size Exclusion Chromatography with Fluorescence Detection. Anal. Chem. 2016, 88, 10390–10394. [Google Scholar] [CrossRef]
- Sidhom, K.; Obi, P.O.; Saleem, A. A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef]
- Turner, N.P.; Abeysinghe, P.; Kwan Cheung, K.A.; Vaswani, K.; Logan, J.; Sadowski, P.; Mitchell, M.D. A Comparison of Blood Plasma Small Extracellular Vesicle Enrichment Strategies for Proteomic Analysis. Proteomes 2022, 10, 19. [Google Scholar] [CrossRef]
- Rider, M.A.; Hurwitz, S.N.; Meckes, D.G., Jr. ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep. 2016, 6, 23978. [Google Scholar] [CrossRef]
- García-Romero, N.; Madurga, R.; Rackov, G.; Palacín-Aliana, I.; Núñez-Torres, R.; Asensi-Puig, A.; Carrión-Navarro, J.; Esteban-Rubio, S.; Peinado, H.; González-Neira, A.; et al. Polyethylene glycol improves current methods for circulating extracellular vesicle-derived DNA isolation. J. Transl. Med. 2019, 17, 75. [Google Scholar] [CrossRef]
- Ludwig, K.; Miroschedji, K.D.; Doeppner, T.R.; Börger, V.; Ruesing, J.; Rebmann, V.; Durst, S.; Jansen, S.; Bremer, M.; Behrmann, E.; et al. Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. J. Extracell. Vesicles 2018, 7, 1528109. [Google Scholar] [CrossRef]
- Pavani, K.C.; Lin, X.; Hamacher, J.; Broeck, W.V.D.; Couck, L.; Peelman, L.; Hendrix, A.; Van Soom, A. The separation and characterization of extracellular vesicles from medium conditioned by bovine embryos. Int. J. Mol. Sci. 2020, 21, 2942. [Google Scholar] [CrossRef]
- Keller, B.O.; Sui, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Lucien, F.; Gustafson, D.; Lenassi, M.; Li, B.; Teske, J.J.; Boilard, E.; Clemm con Hohenberg, K.; Falcón-Perez, H.M.; Gualerzi, A.; Reale, A.; et al. MIBlood-EV: Minimal information to enhance the quality and reproducibility of blood extracellular vesicle research. J. Extracell. Vesicles 2023, 12, e12385. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, R.; Siljander, P.R. A beginner’s guide to study extracellular vesicles in human blood plasma and serum. J. Extracell. Vesicles 2024, 13, e12400. [Google Scholar] [CrossRef]
- Lim, H.J.; Yoon, H.; Kim, H.; Kang, Y.W.; Kim, J.E.; Kim, O.Y.; Lee, E.Y.; Twizere, J.C.; Rak, J.; Kim, D.K. Extracellular Vesicle Proteomes Shed Light on the Evolutionary, Interactive, and Functional Divergence of Their Biogenesis Mechanisms. Front. Cell Dev. Biol. 2021, 9, 734950. [Google Scholar] [CrossRef]
- Greening, D.W.; Xu, R.; Gopal, S.K.; Rai, A.; Simpson, R.J. Proteomic insights into extracellular vesicle biology—Defining exosomes and shed microvesicles. Expert Rev. Proteom. 2017, 14, 69–95. [Google Scholar] [CrossRef]
- Mallia, A.; Gianazza, E.; Zoanni, B.; Brioschi, M.; Barbieri, S.S.; Banfi, C. Proteomics of Extracellular Vesicles: Update on Their Composition, Biological Roles and Potential Use as Diagnostic Tools in Atherosclerotic Cardiovascular Diseases. Diagnostics 2020, 10, 843. [Google Scholar] [CrossRef]
- Katsuda, T.; Kosaka, N.; Ochiya, T. The roles of extracellular vesicles in cancer biology: Toward the development of novel cancer biomarkers. Proteomics 2014, 14, 412–425. [Google Scholar] [CrossRef]
- Vanderboom, P.M.; Dasari, S.; Ruegsegger, G.N.; Pataky, M.W.; Lucien, F.; Heppelmann, C.J.; Lanza, I.R.; Nair, K.S. A size-exclusion-based approach for purifying extracellular vesicles from human plasma. Cell Rep. Methods 2021, 1, 100055. [Google Scholar] [CrossRef]
- Vallejo, M.C.; Sarkar, S.; Elliott, E.C.; Henry, H.R.; Powell, S.M.; Lucovico, I.D.; You, Y.; Huang, F.; Payne, S.H.; Ramanadham, S.; et al. A proteomic meta-analysis refinement of plasma extracellular vesicles. Sci. Data 2023, 10, 837. [Google Scholar] [CrossRef]
- Braun, F.; Rinschen, M.; Buchner, D.; Bohl, K.; Plagmann, I.; Bachurski, D.; Späth, M.R.; Antczak, P.; Göbel, H.; Klein, C.; et al. The proteomic landscape of small urinary extracellular vesicles during kidney transplantation. J. Extracell. Vesicles 2020, 10, e12026. [Google Scholar] [CrossRef]
- Vallabhajosyula, P.; Korutla, L.; Habertheuer, A.; Yu, M.; Rostami, S.; Yuan, C.-X.; Reddy, S.; Liu, C.; Korutla, V.; Koeberlein, B.; et al. Tissue-specific exosome biomarkers for noninvasively monitoring immunologic rejection of transplanted tissue. J. Clin. Investig. 2017, 127, 1375–1391. [Google Scholar] [CrossRef] [PubMed]
- Chettimada, S.; Lorenz, D.R.; Misra, V.; Dillon, S.T.; Reeves, R.K.; Manickam, C.; Morgello, S.; Kirk, G.D.; MEhta, S.H.; Gabuzda, D. Exosome markers associated with immune activation and oxidative stress in HIV patients on antiretroviral therapy. Sci. Rep. 2018, 8, 7227. [Google Scholar] [CrossRef] [PubMed]
- Buenafe, A.C.; Dorrell, C.; Reddy, A.P.; Klimek, J.; Marks, D.L. Proteomic analysis distinguishes extracellular vesicles produced by cancerous versus healthy pancreatic organoids. Sci. Rep. 2022, 12, 3556. [Google Scholar] [CrossRef]
- Ialongo, C.; Bernardini, S. Phlebotomy, a bridge between laboratory and patient. Biochem. Med. (Zagreb) 2016, 26, 17–33. [Google Scholar] [CrossRef]
- Baranyai, T.; Herczeg, K.; Onódi, Z.; Voszka, I.; Módos, K.; Marton, N.; Nagy, G.; Mäger, I.; Wood, M.J.; El Andaloussi, S.; et al. Isolation of Exosomes from Blood Plasma: Qualitative and Quantitative Comparison of Ultracentrifugation and Size Exclusion Chromatography Methods. PLoS ONE 2015, 10, e0145686. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; McGee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci Rep. 2020, 10, 1039. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, e23430. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef]
- Yuana, Y.; Böing, A.N.; Grootemaat, A.E.; van der Pol, E.; Hau, C.M.; Cizmar, P.; Buhr, E.; Sturk, A.; Nieuwland, R. Handling and storage of human body fluids for analysis of extracellular vesicles. J. Extracell. Vesicles 2015, 4, e29260. [Google Scholar] [CrossRef] [PubMed]
- Kabagwira, J.; Gonda, A.; Vallejos, P.; Babcock, B.; Langridge, W.H.R.; Senthil, M.; Wall, N.R. Optimization of blood handling for plasma extracellular vesicle isolation. J. Nanopart. Res. 2021, 23, 65. [Google Scholar] [CrossRef]
- Lacroix, R.; Judicone, C.; Poncelet, P.; Robert, S.; Arnaud, L.; Sampol, J.; Dignat-George, F. Impact of pre-analytical parameters on the measurement of circulating microparticles: Towards standardization of protocol. J. Thromb. Haemost. 2012, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Fliser, E.; Jerkovic, K.; Vidovic, T.; Gorenjak, M. Investigation of unusual high serum indices for lipemia in clear serum samples on siemens analysers dimension. Biochem. Med. (Zagreb) 2012, 22, 352–362. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Pálóczi, K.; Kovács, A.; Barabás, E.; Bekó, G.; Várnai, K.; Pállinger, É.; Szabó-Taylor, K.; Szabó, T.G.; Kiss, A.A.; et al. Improved circulating microparticle analysis in acid-citrate dextrose (ACD) anticoagulant tube. Thromb. Res. 2014, 133, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, L.S.; Santana, M.M.; Henriques, C.; Pinto, M.M.; Ribeiro-Rodrigues, T.M.; Girão, H.; Nobre, R.J.; Pereira de Almeida, L. Simple and Fast SEC-Based Protocol to Isolate Human Plasma-Derived Extracellular Vesicles for Transcriptional Research. Mol. Ther. Methods Clin. Dev. 2020, 18, 723–737. [Google Scholar] [CrossRef]
- Vasco, F.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Koncz, A.; Turiák, L.; Németh, K.; Lenzinger, D.; Bárkai, T.; Lőrincz, P.; Zelenyánszki, H.; Vukman, K.V.; Buzás, E.I.; Visnovitz, T. Endoplasmin Is a Hypoxia-Inducible Endoplasmic Reticulum-Derived Cargo of Extracellular Vesicles Released by Cardiac Cell Lines. Membranes 2023, 13, 431. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Turiák, L.; Misják, P.; Szabó, T.G.; Aradi, B.; Pálóczi, K.; Ozohanics, O.; Drahos, L.; Kittel, A.; Falus, A.; Buzás, E.I.; et al. Proteomic characterization of thymocyte-derived microvesicles and apoptotic bodies in BALB/c mice. J. Proteom. 2011, 74, 2025–2033. [Google Scholar] [CrossRef]
- Bugyi, F.; Tóth, G.; Kovács, K.B.; Drahos, L.; Turiák, L. Comparison of solid-phase extraction methods for efficient purification of phosphopeptides with low sample amounts. J. Chromatogr. A 2022, 1685, 463597. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinform. 2012, 13, 13.20.1–13.20.14. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Zhen, K.; Wei, X.; Zhi, Z.; Zhang, S.; Cui, L.; Li, Y.; Chen, X.; Yao, J.; Zhang, H. Comparison of Different Isolation Methods for Plasma-Derived Extracellular Vesicles in Patients with Hyperlipidemia. Life 2022, 12, 1942. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Boilard, E.; Buzas, E.I.; Cheng, L.; Falcón-Perez, J.M.; Gardiner, C.; Gustafson, D.; Gualerzi, A.; Hendrix, A.; Hoffman, A.; et al. Considerations towards a roadmap for collection, handling and storage of blood extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1647027. [Google Scholar] [CrossRef]
- Bettin, B.; Gasecka, A.; Li, B.; Dhondt, B.; Hendrix, A.; Nieuwland, R.; Van der Pol, E. Removal of platelets from blood plasma to improve the quality of extracellular vesicle research. J. Thromb. Haemost. 2022, 20, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Dhondt, B.; Pinheiro, C.; Geeurickx, E.; Tulkens, J.; Vergauwen, G.; Van Der Pol, E.; Nieuwland, R.; Decock, A.; Miinalainen, I.; Rappu, P.; et al. Benchmarking blood collection tubes and processing intervals for extracellular vesicle performance metrics. J. Extracell. Vesicles 2023, 12, e12315. [Google Scholar] [CrossRef] [PubMed]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Ter-Ovanesyan, D.; Norman, M.; Lazarovits, R.; Trieu, W.; Lee, J.H.; Church, G.M.; Walt, D.R. Framework for rapid comparison of extracellular vesicle isolation methods. eLife 2021, 10, e70725. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rhodes, M.; Adlou, B.; Williams, S.; Lees, R.; Peacock, B.; Aubert, D.; Jalal, A.R.; Lewis, M.P.; Davies, O.G. Defining the influence of size-exclusion chromatography fraction window and ultrafiltration column choice on extracellular vesicle recovery in a skeletal muscle model. J. Extracell. Biol. 2023, 2, e85. [Google Scholar] [CrossRef]
- Ekström, K.; Crescitelli, R.; Pétursson, H.I.; Johansson, J.; Lässer, C.; Bagge, R.O. Characterization of surface markers on extracellular vesicles isolated from lymphatic exudate from patients with breast cancer. BMC Cancer 2022, 22, 50. [Google Scholar] [CrossRef]
- Sódar, B.W.; Kittel, Á.; Pálóczi, K.; Vukman, K.V.; Osteikoetxea, X.; Szabó-Taylor, K.; Németh, A.; Sperlágh, B.; Baranyai, T.; Giricz, Z.; et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci. Rep. 2016, 6, 24316. [Google Scholar] [CrossRef]
- Buzás, E.I.; Tóth, E.Á.; Sódar, B.W.; Szabó-Taylor, K.É. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef]
- Veerman, R.E.; Teeuwen, L.; Czarnewski, P.; Güclüler Akpinar, G.; Sandberg, A.; Cao, X.; Pernemalm, M.; Orre, L.M.; Gabrielsson, S.; Eldh, M. Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. J. Extracell. Vesicles 2021, 10, e12128. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.; Coumans, F.A.; Maltesen, R.G.; Böing, A.N.; Bonnington, K.E.; Broekman, M.L.; Broom, M.F.; Buzás, E.I.; Christiansen, G.; Hajji, N.; et al. A standardized method to determine the concentration of extracellular vesicles using tunable resistive pulse sensing. J. Extracell. Vesicles 2016, 5, 31242. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
György, B.; Pálóczi, K.; Balbisi, M.; Turiák, L.; Drahos, L.; Visnovitz, T.; Koltai, E.; Radák, Z. Effect of the 35 nm and 70 nm Size Exclusion Chromatography (SEC) Column and Plasma Storage Time on Separated Extracellular Vesicles. Curr. Issues Mol. Biol. 2024, 46, 4337-4357. https://doi.org/10.3390/cimb46050264
György B, Pálóczi K, Balbisi M, Turiák L, Drahos L, Visnovitz T, Koltai E, Radák Z. Effect of the 35 nm and 70 nm Size Exclusion Chromatography (SEC) Column and Plasma Storage Time on Separated Extracellular Vesicles. Current Issues in Molecular Biology. 2024; 46(5):4337-4357. https://doi.org/10.3390/cimb46050264
Chicago/Turabian StyleGyörgy, Bernadett, Krisztina Pálóczi, Mirjam Balbisi, Lilla Turiák, László Drahos, Tamás Visnovitz, Erika Koltai, and Zsolt Radák. 2024. "Effect of the 35 nm and 70 nm Size Exclusion Chromatography (SEC) Column and Plasma Storage Time on Separated Extracellular Vesicles" Current Issues in Molecular Biology 46, no. 5: 4337-4357. https://doi.org/10.3390/cimb46050264