

Atomistic Details of Methyl Linoleate Pyrolysis: Direct Molecular Dynamics Simulation of Converting Biodiesel to Petroleum Products


Department of Chemistry & Biochemistry, Missouri State University, Springfield, MO 65897, USA
*
Author to whom correspondence should be addressed.
†
Present Address: Department of Chemistry, Charles University, Akademika Heyrovského 1203/8, 500 05 Hradec Králové, Czech Republic.
Energies 2024, 17(10), 2433; https://doi.org/10.3390/en17102433
Submission received: 9 April 2024
/
Revised: 25 April 2024
/
Accepted: 16 May 2024
/
Published: 20 May 2024
(This article belongs to the Special Issue Biodiesel and Biolubricant: Production, Sources and Environmental Impact)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Dependence on petroleum and petrochemical products is unsustainable; it is both a finite resource and an environmental hazard. Biodiesel has many attractive qualities, including a sustainable feedstock; however, it has its complications. The pyrolysis (a process already in common use in the petroleum industry) of biodiesel has demonstrated the formation of smaller hydrocarbons comprising many petrochemical products but experiments suffer from difficulty quantifying the myriad reaction pathways followed and products formed. A computational simulation of pyrolysis using “ab initio molecular dynamics” offers atomic-level detail of the reaction pathways and products formed. Herein, the most prevalent fatty-acid ester (methyl linoleate) from the most prevalent feedstock for biodiesel in the United States (soybean oil) is studied. Temperature acceleration within the atom-centered density matrix propagation formalism (Car–Parrinello) utilizing the D3-M06-2X/6-31+G(d,p) model chemistry is used to compose an ensemble of trajectories. The results are grounded in comparison to experimental studies through agreement in the following: (1) the extent of reactivity (40% in the experimental and 36.1% in this work), (2) the homology of hydrocarbon products formed (wt % of C6–C10 products), and (3) the CO/CO2 product ratio. Deoxygenation pathways are critically analyzed (as the presence of oxygen in biodiesel represents a disadvantage in its current use). Within this ensemble, deoxygenation was found to proceed through two subclasses: (1) spontaneous deoxygenation, following one of four possible pathways; or (2) induced deoxygenation, following one of three possible pathways.
1. Introduction
Petroleum and petrochemical products are relevant to a wide variety of industries including medicine [1], research, manufacturing, energy, and transportation [2,3]. However, environmental concerns and depleting reserves [2] have society clamoring for alternatives. Many technologies of interest show great potential for remediating the energy crisis, though they often fall short of completely replacing petroleum due to our rampant use of petrochemical products [4]. For example, much of the polymer industry is reliant on petroleum for precursor olefins [5], although research is quickly expanding in the area of biologically derived polymers for, e.g., the medical industry. Despite the massive success of some of these alternatives, obtaining energy from natural sources such as solar, wind, geothermal, or hydroelectric will never obviate the need for olefins currently obtained from petroleum. Without significant sources of massive quantities of olefins [6,7], polymer supply chains will be massively disrupted, affecting the market, economy, scientific research, and healthcare [1].
Biodiesel is an alternative energy source currently implemented on a relatively small scale as a transportation (diesel) fuel alternative, but it may also hold the key to producing petrochemical products as well. Biodiesel, a subclass of biofuels, is derived from natural sources [8] such as vegetable oils [9,10,11] or animal fats (triglycerides) [12]. Vegetable oil triglycerides offer a more sustainable feedstock; the variety of vegetable oil used varies depending on the local climate [13]. In the US, soybean oil is currently the most common vegetable oil utilized for biodiesel, while in Europe, canola oil is more prominent [8,14]. Regardless of the triglyceride used, transesterification is employed (Figure 1), usually with methanol, to create fatty-acid methyl esters (FAMEs, as well as glycerol). Utilization of FAMEs in a combustion engine has two major advantages: (1) it can be directly used in combustion engines without modification [14,15], and, (2) biosynthetically, it is created by photosynthesis incorporating carbon dioxide, making the process essentially carbon neutral [4,14,16]. In addition to remediating our release of greenhouse gases, the burning of FAMEs and biodiesel has shown significantly reduced rates of nitrogen oxides (NOx) being emitted. Switching to a domestic energy source also has the potential to improve market stability and reinforce the agricultural industry by introducing a targeted high-value crop [14,16,17].
There are many obstacles restricting widespread implementation of biodiesel in diesel engines [18]. While emissions of sulfur oxides (SOx) are reduced, NOx emissions rise in comparison to fossil fuels [4,14,16]. Additionally, FAMEs are amphiphilic and act as surfactants, which has been shown to cause damage to engines [19]. Modified exhaust filters are available, though added costs would detract from their economic appeal. The addition of hydrogen gas to the biodiesel fuel during combustion (duel fuel) has been shown to improve emissions, including NOx [20,21] but requires engine retrofitting. Biodiesel also costs approximately one and a half times as much to produce compared to its petroleum-based counterpart [12,22]. Yet another complication associated with biodiesel is its operability in colder climates. Operability designates the minimum conditions a fuel source is usable. The typical pour point (the point at which gel forms) is higher than the typical diesel product (as high as 15 °C) [15,19]. Improving biodiesel’s low-temperature operability would be significant for improving its efficacy [19,23]. The poor low-temperature operability and surfactant properties are due to the homology and polar ester group; removing the polar ester group and reducing the length of hydrocarbon chains in biodiesel would facilitate its implementation [17]. The pyrolysis of biodiesel would address both issues.
The result of pyrolysis on hydrocarbons is the creation of new smaller hydrocarbons with many improved properties, and it is already readily available and employed in the petroleum industry. The application of this technique to biofuels has been shown capable of producing similar hydrocarbons to that of petroleum, as well as many of the by-products essential for the replacement of petroleum in society [24]. Efforts to improve this process have been conducted for both industrial and academic pursuits to give the technique more widespread appeal [25]. Published works on FAMEs (mixtures and pure) have investigated ideal conditions (e.g., temperature/pressure) for producing shorter-chain hydrocarbons from oleic [26], linoleic, and stearic [27] acids [15]. Other investigations include microwave-assisted [28] and flash [29] pyrolysis on FAME precursors (triglycerides) in biomass and castor oil with positive results. Promising results have also been obtained from introducing activated alumina to the process [30]. While the findings of these studies show tremendous potential, a limitation in the research is that pyrolysis reactions are difficult to control or measure. Additionally, the costs of equipment and apparatus can make experimental investigations cost-restrictive [13,14].
Theoretical chemists have also taken up FAME and triglyceride pyrolysis in various capacities. Electronic structure theory has been applied to investigate bond dissociation energies (BDEs) of the FAMEs methyl linolenate [31], as well as methyl linoleate and methyl oleate [19]. These studies utilized density functional theory (DFT) to complete electronic structure calculations for the myriad bond dissociations possible in each of these FAMEs. To a large degree, these studies confirm predictions often made by chemists. Namely, that C–C bonds, that would break to form allylic radicals, are the most likely points for bond breakage, with select C–H bonds (that would also produce allylic radicals) a close second.
Reactive force field (ReaxFF)-based molecular dynamics have been employed for investigating inter- and intramolecular interactions to determine fuel properties [32]. ReaxFF methods incorporate elements of quantum chemistry into parameterization of potentials. This results in a tremendous speed-up of molecular dynamics calculations but leaves questions as to whether it might be better suited for interpolation. This is to be distinct from extrapolation, where species are created that were not part of the parameterization set. To our knowledge, no ‘direct dynamics’ investigation into the propagation of pyrolysis with biodiesel exists. The major advantage of direct dynamics over ReaxFF techniques is the inclusion of direct quantum calculations into the dynamics in a manner that is limited only to the caveats of the model chemistry.
Variations among FAMEs present in biodiesel are a function of the triglyceride and, by extension, the form of feedstock used. FAMEs differ in homology (length) and saturation (presence, location, and configuration of alkenes) [33,34]. The differences exhibited between FAMEs are of significant interest as they relate directly to fuel quality in biodiesel. FAME derived from soybean oil—the most common feedstock currently used for biodiesel production in the United States of America—contains high concentrations of methyl linoleate (C18:2—54.5%) and methyl oleate (C18:1—24.1%). Europe’s predominant feedstock for current biodiesel production, canola (rapeseed) oil, produces 19.1–20.0 wt % methyl linoleate and 60.0–64.3 wt % methyl oleate [8]. The focus herein will be on methyl linoleate (Figure 2), the largest component of US-derived biodiesel and the second-largest component of European-derived biodiesel.
This study seeks an atomic-level understanding of the pyrolysis of methyl linoleate. Comparisons between the ensemble assembled herein and experimental studies require statistical analysis of the data points obtained. For a trajectory product to be considered “significant”, the frequency of occurrence must be higher than the deviation caused by error bias. These error calculations are derived not from the method but by limitations imposed by the size of the ensemble (100).
2. Methods
Herein, direct dynamics simulations that integrate Newton’s classical equations of motion with forces derived from an electronic structure method. The Gaussian09 [35] suite of programs was used to compute all trajectories.
2.1. Electronic Structure Calculations and Model for the Potential Energy Surface
Our previous work in evaluating the accuracy of a model chemistry, tempered by its computational cost (as applied to FAMEs), showed that unrestricted M06-2X/6-31+G(d,p) reduced the error (in BDE) to 2.8% while maintaining a tractable computational cost [19]. The M06-2X functional was formulated by Truhlar and Zhao to describe organic molecular systems. Specifically parameterized to capably simulate π–π interactions, hydrogen bonding, and long-range forces; the method has been utilized recently in a benchmark BDE investigation for methyl linolenate [36]. Through investigation, the method was found to accurately describe the thermochemistry, kinetics, and non-covalent interactions of the main block elements [36].
2.2. Computational Details of the Direct Dynamics Simulations
Two typical methods are utilized for direct molecular dynamics, the Born–Oppenheimer molecular dynamics (BOMD) approach and the extended Lagrangian molecular dynamics (ELMD) approach [41]. Both methods can accurately simulate different reaction dynamics, but each has its own advantages and disadvantages. BOMD derives information about the potential energy surface through de novo convergence of an electronic structure calculation. This method of system propagation is extremely accurate and can describe details of the quantum model to an excellent degree. Unfortunately, due to the expensive nature of electronic structure calculations, the computational cost adds up quickly. ELMD offers an elegant solution to this problem by propagating the wavefunction along with the classical nuclear degrees of freedom. This is achieved by introducing Kohn–Sham or Gaussian orbitals in a plane wave basis set approach and adjusting the relative time scales of electronic and nuclear motion. It has been discovered that employing atom-centered density matrix propagation (ADMP) can effectively describe the dynamics for cluster- and gas-phase reactions. Additionally, the scaling of ADMP is based on the DFT linear scaling, O(N), giving it the capability to describe larger systems, such as that discussed in this paper.
ADMP has been used to describe the chemical nature of fairly large systems while still offering rigorous treatment of all electrons. It does this by treating electrons with pseudopotentials, while still permitting reasonably large time steps through the incorporation of tensorial fictitious masses. It can also be employed effectively with exchange–correlation functionals, including the hybrid density functional discussed above. Additionally, and extremely pertinent to the scope of this research, ADMP allows for rigorous ‘on the fly’ deviations from the Born–Oppenheimer surface, which is necessary to produce a stochastic ergodic ensemble.
Experimental investigations of pyrolysis indicate timescales of 4–8 h, while direct dynamics simulations realistically comprise tens of ps. To remediate this gap, temperature-accelerated molecular dynamics (TAMD) was implemented [42].
TAMD is typically instituted for describing systems with slow metastable state-to-state transitions as compared to thermal vibrations [42]. The ability to describe infrequent (rare) events is extremely useful for many MD investigations, including describing protein folding [43], bulk and surface diffusion processes [44], or large-scale systems and reaction mechanisms [45], such as that presented in this study. Assuming harmonic transition state theory, TAMD permits simulations for long-time dynamics consisting of infrequent jumps between states that cannot be completely mapped out [46]. Through increasing conformational and constitutional changes [47] trajectories obtained using this method no longer remain close to only one minimum (presumably the initial state). Choosing the proper temperature to accelerate the system is an important consideration in investigations like these. If the temperature is set too high, unphysical states may be accessed during the simulation (as compared to hours at a lower temperature). Herein, the temperature was set to 3500 K, which showed no unphysical behavior (vide infra) while providing a reasonable rate of dissociation (100 of 277). Trajectories were configured to simulate 2.000 ps in 1.0 fs time steps [48,49].
Trajectories were initialized from a single conformation of methyl linoleate, previously determined [19] with a pseudo-random distribution of nuclear kinetic energy (NKE), as computed by Equation (1) [50]. The NKE was monitored throughout the trajectory (every 10 steps) and adjusted if the temperature deviated by more than 2.0 K from the specified temperature of 3500 K. The self-consistent field (SCF) equations were fully converged at each step of the trajectory with spin symmetry broken [utilizing guess = (mix,always)].
3. Results and Discussion
A total of 100 trajectories exhibiting bond dissociation were collected (277 total trajectories were run to acquire these data, i.e., 36.1% of trajectories showed dissociation). Although configured to run 2.0 ps (2000 fs), the average trajectory stopped at 1189 fs of simulated time. The overall distribution of the reactive trajectories is displayed in Figure 3 by plotting the time versus inter-atomic distance for the pair of atoms that represent the first dissociation for each molecule in the ensemble. While the number of bonds broken in each trajectory varies from one to five, within the allotted time, only a third of the jobs show a single dissociation. The average number of bonds broken in each trajectory is 2.27 (vide infra). Overall, the observed product distribution was highly varied, although many products were observed many times, e.g., ethene was produced 57 times within the ensemble. Other products appeared in high frequencies but with some small degree of augmentation (e.g., several substituted cyclopropanes were observed). Further analysis of the data is reliant on an ensemble analysis, micro-scale product analysis, and atomic-level mechanism interpretation.
3.1. Ensemble Analysis
Reactions occurring in pyrolysis are heavily free-radical-dominated mechanisms [51,52] and products are often characterized experimentally by mass spectrometry (MS) and/or gas chromatography (GC) [26,27,34]. Herein, such experimental data are used to assess the ensemble of trajectories compiled (see Supplementary Material for structures produced in each reactive trajectory). Previous theoretical reports, namely bond dissociation energy (BDE) calculations, also offer a point to assess the ensemble assembled herein. Thus, three categories of comparison are discussed herein: the homology of hydrocarbon products, the ratio of CO2 to CO production, and the alignment of ensemble results and bond dissociation energy analysis.
3.2. Homology of Hydrocarbon Products
A report by Asomaning et al. interpreted the distribution of products based on the thermal decomposition of FAMEs (oleic and linoleic) [34]. Products observed in the trajectory simulations were binned according to the number of carbons (Figure 4a; see Supplementary Material for summary of products by mass and homolgy) and compared to those reported by Asomaning et al. (Figure 4b). The experimental work included runs at multiple temperatures from 350–450 °C. Of note is the fact that the data obtained theoretically seem to match most closely with the experimental data obtained at 350 °C. This can be seen in comparing the wt % of 6-carbon products formed in each of the theoretical and experimental plots, as well as the general shape of the decay in proceeding from 6- to 10-carbon products. Also, note that the wt % of the 6-carbon products from the ensemble herein is not a match for the wt % at any of the higher experimental temperatures.
3.3. Ratio of CO2 to CO Production
FAME pyrolysis results in decarboxylation and decarbonylation. Experimentally, the quantity of CO2 and CO have been measured as a function of temperature, see Figure 5. Comparing these results to the quantity obtained theoretically (2.8 ± 0.2% CO2, 1.87 ± 0.6% CO, 1:1.497), this seems to agree with the previous observation of lower temperature pyrolysis (<350 °C).
The experimentally determined CO and CO2 production is affected by post-pyrolysis reactions, as noted by Asomaning et al. [34], where water or methanol react during the cool-down phase of the experiment. For example, a gas-shift reaction is known to occur post-pyrolysis that increases the proportion of H2 and CO2 observed. It is important to note that the simulations discussed herein aim to model the unimolecular reaction dynamics and do not account for post-pyrolysis cool-down/derivatization.
3.4. Alignment of Ensemble Results and Bond-Dissociation Energy (BDE) Analysis
Electronic structure calculations have been used to compute accurate BDEs for methyl linoleate [19], among other FAMEs [31]. Although these electronic structure methods produce accurate values for the thermochemistry involved in bond dissociation, they describe initial bond dissociation. Therefore, any phenomena resulting from the redistribution of energy (including, e.g., subsequent bond dissociation) represents a separate paradigm not considered. In Figure 6 the BDE for each carbon–carbon bond (red, solid bars) is plotted vertically above the frequency of that bond dissociating in the ensemble (blue, hashed bars; see Supplementary Material for the observed frequency for each C-C bond breaking).
The general trends displayed in Figure 6 suggest consistency between the BDE calculations [19] and the results of the direct dynamics simulations [42]. For example, bonds with exceedingly high BDEs (~160 kcal/mol) are unlikely to dissociate (this includes C9–C10 and C12–C13 shown in Figure 6, but also C1–Ob bond cleavage not shown in the Figure). Those C–C bonds with the lowest BDEs include those to an allylic position (C7–C8 and C14–C15) and these are found to dissociate more frequently in the ensemble. Homolysis of the C10–C11 and C11–C12 result in allylic radicals, however, they simultaneously form vinyl radical structures; the instability of these vinyl radicals is more easily noticed in the C8–C9 and C10–C11 bonds that exhibit comparatively larger BDEs (and correspondingly lower frequency of dissociation occurrences).
There are also somewhat surprising deviations exhibited in Figure 6. For example, the C1–C2 bond is observed to break somewhat frequently, despite its BDE being near the top value for C–C single bonds in methyl linoleate. One quickly notices that this is logical, when considering that the major advantage to the dynamics simulations (over BDE analysis) is that subsequent dissociations are modeled: cleavage of the C1–C2 bond leads to decarboxylation—a notable chemical sink. Also, note that this assertion is consistent with the experimental findings and the fact that CO2 gas is detected (vide supra).
The bonds between C2–C3, C6–C7, C15–C16, and C17–C18 all show low frequencies of dissociation in the dynamics simulations despite their relatively low BDE (comparable to, e.g., C6–C7 and C16–C17). Notably, cleavage of C6–C7 and C16–C17 lead to even-numbered hydrocarbon radicals that undergo β-scission to form compounds, including ethylene. Cleavage at C2–C3, C6–C7, C15–C16, and C17–C18 frustrates this process. Herein, this process is described as a “zipper” effect or “β-scission cascade” (Figure 7), comparable to β-oxidation in biological fatty-acid metabolism.
The end product of “β-scission cascade” is ethylene, a vital precursor for many industrial reactions. Several ketenes are also formed in the trajectories, the simplest being ethenone. While useful, this gas is so reactive in situ, an immediate acetylation will occur resulting in either amides, esters, carboxylic acids, or acid anhydrides.
Cyclization was observed in both theoretical and experimental results [26,34], though the quantities do not directly relate. This can be explained by the fact that many of the reactions seen in the ensemble constitute initiation-phase free-radical mechanisms, with some propagation. Since cyclization involves bond formation, products of this kind are most likely a result of late-propagation and termination steps, which are not observed in high frequencies within the relatively short time scale of the trajectories in this ensemble.
3.5. Atomic-Level Mechanisms
The final interpretation of the trajectories presented herein cannot be performed directly through experimental means—atomic-level analysis—i.e., reaction mechanisms. The ultimate product for each reactive trajectory can be found in the Supplementary Material, however, each trajectory must be visualized to determine the reaction mechanism. The lifetime of a radical is short and turbulent and any experimental attempts to derive pyrolysis mechanisms for methyl linoleate has been met with difficulty. Previous experimental studies have shown production of CO2 and CO, indicating that deoxygenation and decarboxylation reactions have taken place, but a lack of atomic-level details means that no specific mechanisms have been proposed. The trajectories for the ensemble assembled herein fall into six primary pathways for deoxygenation that fall into two categories (Figure 8). The first category, characterized by deoxygenation occurring as the first dissociation event, includes decarbonylation (1, Figure 8), β-scission cascade (2, Figure 8), decarboxylation (3, Figure 8), and pericyclic deoxygenation (4, Figure 8). The second category of deoxygenation reactions are characterized by those pathways that have preceding free-radical forming steps and include: methyl free-radical deoxygenation (5, Figure 8), α free-radical deoxygenation (6, Figure 8), and β free-radical deoxygenation (7, Figure 8). All products of these pathways were observed in experimental analysis. Formaldehyde (CH2O) can be formed through methyl free-radical deoxygenation (5), decarbonylation (1) and β free-radical deoxygenation (7). Carbon monoxide (CO) can be formed through decarbonylation (1) and β free-radical deoxygenation (7). Carbon dioxide can be formed through either decarboxylation (3), or through a water–gas shift with carbon monoxide (vide supra). Methanol can be produced either through a pericyclic deoxygenation (4) pathway or a termination reaction between a hydrogen and methoxy radical.
The remaining pathways seen in the ensemble assembled herein are found to fall into two categories: C–C dissociation (80.72% of reactions) and C–H dissociation (10.76% of reactions). In cases where multiple dissociations are observed, the pathways are organized by the first dissociative event. Preliminary dissociation of C–C or C–H bonds leaves a free radical within a fragment that can react further by, e.g., C–C β-scissions or C–H β-abstraction. These two follow-up reactions are seen relatively frequently due, in many cases to conjugation-established (in the case of β-abstraction) or closed-shell ethene formation (by β-scission). To present the pathways observed in the ensemble, the location of the first C–C dissociation for each trajectory (presumably rate-limiting) is plotted with its frequency in Figure 9 (red, solid bars). Further, the cumulative sum of the observed bond cleavages for each C–C bond is plotted in Figure 9 (blue, hashed, bars). Of the C–C bond dissociations that initiate the decomposition, the C7–C8 bond breaks first the most frequently (13%), followed by the C14–C15 bond (9%). These breakages form allylic radicals and are in line with predictions made by thermochemical analysis (i.e., BDE analyses).
In terms of C–H dissociation, the influence of an initiation step drastically increases its likelihood of occurring. Only 3% of all trajectories undergo a C–H dissociation as the first step, while 9% of trajectories experience C–H cleavage as a subsequent step. Given that C–H bonds are, on average, 10–20 kcal/mol higher in energy than C–C bonds, it makes sense that the initial breakage of a C–C bond would likely proceed first, and once the high-energy (reactive) radical intermediate is formed, a C–H bond cleavage to form a closed-shell product proceeds through a more-attainable energy barrier.
When tracking the location of the C–H bond breakages observed as the initiation step, one notices that only six sites along the molecule react in this manner: C4–H, C8–H, C12–H, C15–H, C17–H, and C18–H. While C8–H produces an allylic radical, which might be predicted by thermodynamic analysis of methyl linoleate, the remaining produce alkyl (C4–H, C15–H, C17–H, and C18–H) and vinyl (C12–H) radicals, which only make sense once later events play out in the reactivity of the molecule.
Overall, those bonds predicted to break with high frequency (due to relatively small BDEs) were found to break. However, molecular dynamics simulations, like those presented herein, present a more complete picture of how the reaction proceeds past the first radical formed. For example, Figure 10 shows pathways to some of the most observed products within the ensemble (see Supplementary Material for structures produced in each reactive trajectory). Notably, 1,2-divinylcyclopropanes are observed with a remarkably high frequency (both in the ensemble and in the experiments). From the dynamics simulations, these rearrangements are observed to proceed via the rearrangement of an allylic radical with homoconjugation (one intervening sp3-hybridized carbon). These rearrangements are reminiscent of vinylcyclopropane to homoallyl rearrangements (Figure 11a). Furthermore, the divinylcyclopropane observed in this ensemble may access larger rings through a [3,3]-sigmatropic shift, as shown in Figure 11b.
4. Conclusions
The results presented above illustrate that direct dynamics simulations of methyl linoleate pyrolysis can provide insight into the atomic-level mechanisms and ensemble average trends that correlate to the previous experimental work. These TAMD simulations display a CO2:CO ratio that is extrapolated to be in line with experimental pyrolysis at a slightly lower temperature than the experimental investigations. The simulated hydrocarbon homologies obtained match those from Asomaning et al. [26,34] for samples pyrolyzed at a temperature of 350 °C. Further suggesting the validity of TAMD is the fact that no problematic, energy forbidden, states were observed. Overall, direct dynamics simulations—while computationally expensive—have proven themselves valuable relative to static electronic structure calculations. Elucidation of common mechanistic pathways may lead to the understanding of the pyrolysis of biodiesel to an extent where prediction of product distributions may be possible. This holds promise for a time when the engineering of fatty-acid chains (to be incorporated into plant oils) and pyrolysis conditions could be tuned to produce an outcome, i.e., polymer precursors, fine-chemical precursors, or fuel components.
Studies in this research group focus on further understanding of the pyrolysis processes that convert biodiesel into petroleum products. This includes the following: (1) increasing computational efficacy for modeling unimolecular processes occurring on longer time scales, (2) simulations with stochastic initial conformation that will (presumably) require less equilibration time, and (3) applications of the above to other FAMEs of interest.
Overall, pyrolysis (a process already in common use in the petroleum industry) of biodiesel would allow society to make the most of the benefits of biodiesel (namely, sustainability) while eliminating one of its most common problems (surfactant properties induced by the inclusion of polar head groups). Pyrolysis to form gasoline constituents would require no major changes to the infrastructure of gasoline production or consumer use.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/en17102433/s1, products for each reactive trajectory, products with associated frequency of observation, and other data used to produce the figures herein can be found within the supplementary materials (.XLSX).
Author Contributions
Data were collected primarily by M.J.B. under the guidance of M.R.S. The manuscript was written through contributions by M.J.B. and M.R.S. All authors have read and agreed to the published version of the manuscript.
Funding
Missouri Soybean Merchandising Council (MSMC) project number #20-446-21.
Data Availability Statement
Data are contained within the article and individual trajectory outcomes (along with statistical analysis) are available in supplementary materials (.XLSX). Ensemble dataset is available on request from the authors.
Acknowledgments
We gratefully acknowledge the support from the Missouri Soybean Merchandising Council (MSMC), project number #20-446-21. We thank the Missouri State University Graduate College for their Faculty Research Grant and Thesis Research Funding, the Missouri State University College of Natural and Applied Sciences and Department of Chemistry for their support of this research. Additionally, we thank the Missouri State University Office of the Provost and Office of Student Development and Public Affairs for sponsoring monthly faculty writing retreats that provided direct support for the production of this report.
Conflicts of Interest
The authors declare no competing financial interests.
Abbreviations
ADMP, atom-centered density matrix propagation. BDE, bond dissociation energy. BOMD, Born–Oppenheimer molecular dynamics. GC, gas chromatography. DFT, Density Functional Theory. ELMD, extended Lagrangian molecular dynamics. FAME, fatty acid methyl ester. MS, mass spectrometry. NOx, nitrogen oxides. NKE, nuclear kinetic energy. ReaxFF, reactive force field. SCF, self-consistent field. SOx, sulfur oxides. TAMD, time-accelerated molecular dynamics.
References
- Hess, J.; Bednarz, D.; Bae, J.; Pierce, J. Petroleum and Health Care: Evaluating and Managing Health Care’s Vulnerability to Petroleum Supply Shifts. Am. J. Public Health 2011, 101, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Owusu, P.A.; Asumadu-Sarkodie, S. A Review of Renewable Energy Sources, Sustainability Issues and Climate Change Mitigation. Cogent Eng. 2016, 3, 1167990. [Google Scholar] [CrossRef]
- Al-Sharrah, G.; Elkamel, A.; Almanssoor, A. Sustainability Indicators for Decision-Making and Optimisation in the Process Industry: The Case of the Petrochemical Industry. Chem. Eng. Sci. 2010, 65, 1452–1461. [Google Scholar] [CrossRef]
- Candeia, R.A.; Silva, M.C.D.; Carvalho Filho, J.R.; Brasilino, M.G.A.; Bicudo, T.C.; Santos, I.M.G.; Souza, A.G. Influence of Soybean Biodiesel Content on Basic Properties of Biodiesel–Diesel Blends. Fuel 2009, 88, 738–743. [Google Scholar] [CrossRef]
- Namazi, H. Polymers in Our Daily Life. Bioimpacts 2017, 7, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Jasinska-Walc, L.; Bouyahyi, M.; Duchateau, R. Potential of Functionalized Polyolefins in a Sustainable Polymer Economy: Synthetic Strategies and Applications. Acc. Chem. Res. 2022, 55, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Koczoń, P.; Bartyzel, B.; Iuliano, A.; Klensporf-Pawlik, D.; Kowalska, D.; Majewska, E.; Tarnowska, K.; Zieniuk, B.; Gruczyńska-Sękowska, E. Chemical Structures, Properties, and Applications of Selected Crude Oil-Based and Bio-Based Polymers. Polymers 2022, 14, 5551. [Google Scholar] [CrossRef]
- Bouaid, A.; El boulifi, N.; Hahati, K.; Martinez, M.; Aracil, J. Biodiesel Production from Biobutanol. Improvement of Cold Flow Properties. Chem. Eng. J. 2014, 238, 234–241. [Google Scholar] [CrossRef]
- Aboim, J.B.; Oliveira, D.; Ferreira, J.E.; Siqueira, A.S.; Dall’Agnol, L.T.; Rocha Filho, G.N.; Gonçalves, E.C.; Nascimento, L.A. Determination of Biodiesel Properties Based on a Fatty Acid Profile of Eight Amazon Cyanobacterial Strains Grown in Two Different Culture Media. RSC Adv. 2016, 6, 109751–109758. [Google Scholar] [CrossRef]
- Lappi, H.; Alén, R. Production of Vegetable Oil-Based Biofuels—Thermochemical Behavior of Fatty Acid Sodium Salts during Pyrolysis. J. Anal. Appl. Pyrolysis 2009, 86, 274–280. [Google Scholar] [CrossRef]
- Su, G.; Ong, H.C.; Mofijur, M.; Mahlia, T.M.I.; Ok, Y.S. Pyrolysis of Waste Oils for the Production of Biofuels: A Critical Review. J. Hazard. Mater. 2022, 424, 127396. [Google Scholar] [CrossRef] [PubMed]
- Graboski, M.S.; McCormick, R.L. Combustion of Fat and Vegetable Oil Derived Fuels in Diesel Engines. Prog. Energy Combust. Sci. 1998, 24, 125–164. [Google Scholar] [CrossRef]
- Lin, L.; Cunshan, Z.; Vittayapadung, S.; Xiangqian, S.; Mingdong, D. Opportunities and Challenges for Biodiesel Fuel. Appl. Energy 2011, 88, 1020–1031. [Google Scholar] [CrossRef]
- Huang, D.; Zhou, H.; Lin, L. Biodiesel: An Alternative to Conventional Fuel. Energy Procedia 2012, 16, 1874–1885. [Google Scholar] [CrossRef]
- Parvizsedghy, R.; Sadrameli, S.M.; Towfighi Darian, J. Upgraded Biofuel Diesel Production by Thermal Cracking of Castor Biodiesel. Energy Fuels 2016, 30, 326–333. [Google Scholar] [CrossRef]
- Jahirul, M.; Rasul, M.; Chowdhury, A.; Ashwath, N. Biofuels Production through Biomass Pyrolysis—A Technological Review. Energies 2012, 5, 4952–5001. [Google Scholar] [CrossRef]
- Knothe, G. Improving Biodiesel Fuel Properties by Modifying Fatty Ester Composition. Energy Environ. Sci. 2009, 2, 759–766. [Google Scholar] [CrossRef]
- Mohamed, M.; Tan, C.-K.; Fouda, A.; Gad, M.S.; Abu-Elyazeed, O.; Hashem, A.-F. Diesel Engine Performance, Emissions and Combustion Characteristics of Biodiesel and Its Blends Derived from Catalytic Pyrolysis of Waste Cooking Oil. Energies 2020, 13, 5708. [Google Scholar] [CrossRef]
- Wilson, Z.R.; Siebert, M.R. Methyl Linoleate and Methyl Oleate Bond Dissociation Energies: Electronic Structure Fishing for Wise Crack Products. Energy Fuels 2018, 32, 1779–1787. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, H.; Li, Y.; Ye, Y.; Tian, J.; Li, J.; Xu, Y.; Lv, J. Research and Optimization of Hydrogen Addition and EGR on the Combustion, Performance, and Emission of the Biodiesel-Hydrogen Dual-Fuel Engine with Different Loads Based on the RSM. Heliyon 2024, 10, e23389. [Google Scholar] [CrossRef]
- Modabberian, A.; Storm, X.; Shamekhi, A.-M.; Vasudev, A.; Zenger, K.; Hyvönen, J.; Mikulski, M. Low Temperature Combustion Modeling and Predictive Control of Marine Engines. Appl. Sci. 2024, 14, 2033. [Google Scholar] [CrossRef]
- Vasudevan, P.T.; Briggs, M. Biodiesel Production—Current State of the Art and Challenges. J. Ind. Microbiol. Biotechnol. 2008, 35, 421. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Zhu, Y.; Tavlarides, L.L. Effect of Thermal Decomposition on Biodiesel Viscosity and Cold Flow Property. Fuel 2014, 117, 981–988. [Google Scholar] [CrossRef]
- Clark, J.; Farmer, T.; Hunt, A.; Sherwood, J. Opportunities for Bio-Based Solvents Created as Petrochemical and Fuel Products Transition towards Renewable Resources. IJMS 2015, 16, 17101–17159. [Google Scholar] [CrossRef] [PubMed]
- Amber, S.; Kamal, M.A. Production of Hydrocarbons by Catalytic Cracking of Stearic Acid under Atmospheric Pressure for Petrochemical Replacement. Pet. Sci. Technol. 2019, 37, 146–154. [Google Scholar] [CrossRef]
- Asomaning, J.; Mussone, P.; Bressler, D.C. Thermal Deoxygenation and Pyrolysis of Oleic Acid. J. Anal. Appl. Pyrolysis 2014, 105, 1–7. [Google Scholar] [CrossRef]
- Maher, K.D.; Kirkwood, K.M.; Gray, M.R.; Bressler, D.C. Pyrolytic Decarboxylation and Cracking of Stearic Acid. Ind. Eng. Chem. Res. 2008, 47, 5328–5336. [Google Scholar] [CrossRef]
- Yin, C. Microwave-Assisted Pyrolysis of Biomass for Liquid Biofuels Production. Bioresour. Technol. 2012, 120, 273–284. [Google Scholar] [CrossRef]
- Vamvuka, D. Bio-Oil, Solid and Gaseous Biofuels from Biomass Pyrolysis Processes-An Overview. Int. J. Energy Res. 2011, 35, 835–862. [Google Scholar] [CrossRef]
- Boocock, D.G.B.; Konar, S.K.; Glaser, G. The Formation of Petrodiesel by the Pyrolysis of Fatty Acid Methyl Esters Over Activated Alumina. In Progress in Thermochemical Biomass Conversion; Bridgwater, A.V., Ed.; Wiley: Hoboken, NJ, USA, 2001; pp. 1517–1524. ISBN 978-0-632-05533-3. [Google Scholar]
- Li, X.; Xu, X.; You, X.; Truhlar, D.G. Benchmark Calculations for Bond Dissociation Enthalpies of Unsaturated Methyl Esters and the Bond Dissociation Enthalpies of Methyl Linolenate. J. Phys. Chem. A 2016, 120, 4025–4036. [Google Scholar] [CrossRef]
- Pires de Oliveira, I.; Caires, A.R.L. Molecular Arrangement in Diesel/Biodiesel Blends: A Molecular Dynamics Simulation Analysis. Renew. Energy 2019, 140, 203–211. [Google Scholar] [CrossRef]
- Lin, R.; Zhu, Y.; Tavlarides, L.L. Mechanism and Kinetics of Thermal Decomposition of Biodiesel Fuel. Fuel 2013, 106, 593–604. [Google Scholar] [CrossRef]
- Asomaning, J.; Mussone, P.; Bressler, D.C. Pyrolysis of Polyunsaturated Fatty Acids. Fuel Process. Technol. 2014, 120, 89–95. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.A.; Al-Hashimi, N.; Shibl, M.F.; Yoshizawa, K.; El-Nahas, A.M. Thermochemistry and Kinetics of the Thermal Degradation of 2-Methoxyethanol as Possible Biofuel Additives. Sci. Rep. 2019, 9, 4535. [Google Scholar] [CrossRef]
- Lynch, B.J.; Zhao, Y.; Truhlar, D.G. Effectiveness of Diffuse Basis Functions for Calculating Relative Energies by Density Functional Theory. J. Phys. Chem. A 2003, 107, 1384–1388. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Steinmetz, M. Effects of London Dispersion Correction in Density Functional Theory on the Structures of Organic Molecules in the Gas Phase. Phys. Chem. Chem. Phys. 2013, 15, 16031. [Google Scholar] [CrossRef]
- Schmid, E.W.; Zieģelmann, H.; Stumpf, H. The Quantum Mechanical Three-Body Problem: Vieweg Tracts in Pure and Applied Physics; Elsevier Science: Kent, UK, 2014; ISBN 978-1-4831-6078-8. [Google Scholar]
- Wilson, Z.R. Ab Initio Methyl Linoleate Bond Dissociation Energies: First Principles Fishing for Wise Crack Products. Master’s Thesis, Missouri State University, Springfield, MO, USA, 2017. [Google Scholar]
- Abrams, C.F.; Vanden-Eijnden, E. Large-Scale Conformational Sampling of Proteins Using Temperature-Accelerated Molecular Dynamics. Proc. Natl. Acad. Sci. USA 2010, 107, 4961–4966. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.-Q.; Lapelosa, M.; Vanden-Eijnden, E.; Abrams, C.F. Full Kinetics of CO Entry, Internal Diffusion, and Exit in Myoglobin from Transition-Path Theory Simulations. J. Am. Chem. Soc. 2015, 137, 3041–3050. [Google Scholar] [CrossRef]
- Schlick, T. Molecular Dynamics-Based Approaches for Enhanced Sampling of Long-Time, Large-Scale Conformational Changes in Biomolecules. F1000 Biol. Rep. 2009, 1. [Google Scholar] [CrossRef]
- Miron, R.A.; Fichthorn, K.A. Accelerated Molecular Dynamics with the Bond-Boost Method. J. Chem. Phys. 2003, 119, 6210–6216. [Google Scholar] [CrossRef]
- Abrams, C.; Bussi, G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2013, 16, 163–199. [Google Scholar] [CrossRef]
- Almas, Q.L.; Keefe, B.L.; Profitt, T.; Pearson, J.K. Choosing an Appropriate Model Chemistry in a Big Data Context: Application to Dative Bonding. Comput. Theor. Chem. 2016, 1085, 46–55. [Google Scholar] [CrossRef]
- Lakshmanan, S.; Pratihar, S.; Hase, W.L. Direct Dynamics Simulations of the CH2 + O2 Reaction on the Ground- and Excited-State Singlet Surfaces. J. Phys. Chem. A 2019, 123, 4360–4369. [Google Scholar] [CrossRef] [PubMed]
- So/rensen, M.R.; Voter, A.F. Temperature-Accelerated Dynamics for Simulation of Infrequent Events. J. Chem. Phys. 2000, 112, 9599–9606. [Google Scholar] [CrossRef]
- Savage, P.E. Mechanisms and Kinetics Models for Hydrocarbon Pyrolysis. J. Anal. Appl. Pyrolysis 2000, 54, 109–126. [Google Scholar] [CrossRef]
- Lappi, H.; Alén, R. Pyrolysis of Vegetable Oil Soaps—Palm, Olive, Rapeseed and Castor Oils. J. Anal. Appl. Pyrolysis 2011, 91, 154–158. [Google Scholar] [CrossRef]
- Krüger, S.; Gaich, T. Recent Applications of the Divinylcyclopropane–Cycloheptadiene Rearrangement in Organic Synthesis. Beilstein J. Org. Chem. 2014, 10, 163–193. [Google Scholar] [CrossRef]
Figure 1.
Reaction scheme describing the transesterification reaction that converts triglycerides into FAMEs and glycerol using sodium hydroxide and methanol.
Figure 1.
Reaction scheme describing the transesterification reaction that converts triglycerides into FAMEs and glycerol using sodium hydroxide and methanol.

Figure 2.
Methyl Linoleate structure. Numbering of the ester begins with the carbonyl carbon and continues down the fatty-acid chain. The methyl carbon will be referred to as “M” and the oxygens as “Oa” and “Ob”.
Figure 2.
Methyl Linoleate structure. Numbering of the ester begins with the carbonyl carbon and continues down the fatty-acid chain. The methyl carbon will be referred to as “M” and the oxygens as “Oa” and “Ob”.

Figure 3.
Interatomic distance for atom pairs representing the first bond dissociation for each trajectory in the ensemble.
Figure 3.
Interatomic distance for atom pairs representing the first bond dissociation for each trajectory in the ensemble.
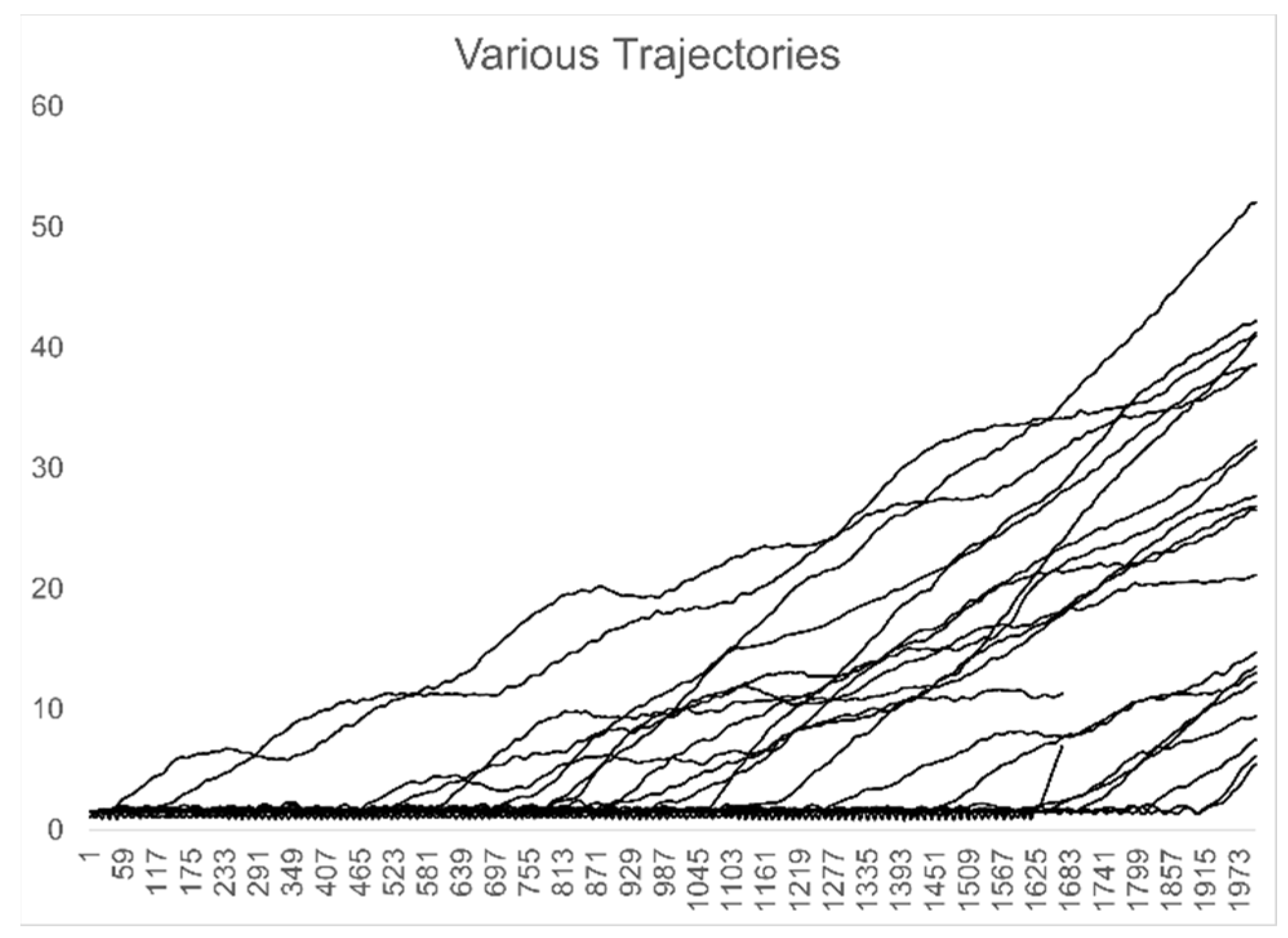
Figure 4.
Comparison between theoretical and experimental results showing the correlation between pyrolysis reactions achieved at 350 °C and those obtained using theoretical means [34].
Figure 4.
Comparison between theoretical and experimental results showing the correlation between pyrolysis reactions achieved at 350 °C and those obtained using theoretical means [34].

Figure 5.
Experimental CO and CO2 production (top). Experimental CO/CO2 ratio (bottom) displays parabolic shape, indicating that theoretical results fit at either a higher or lower temperature than the experiment [34].
Figure 5.
Experimental CO and CO2 production (top). Experimental CO/CO2 ratio (bottom) displays parabolic shape, indicating that theoretical results fit at either a higher or lower temperature than the experiment [34].

Figure 6.
Comparison of theoretical results obtained from benchmarking the BDE investigation and direct dynamics trajectory analysis. Only C–C bonds are shown, as they are the most relevant to the discussion of pyrolysis chemistry [42].
Figure 6.
Comparison of theoretical results obtained from benchmarking the BDE investigation and direct dynamics trajectory analysis. Only C–C bonds are shown, as they are the most relevant to the discussion of pyrolysis chemistry [42].

Figure 7.
While initiation can occur at specific weak sites within the molecule, the “zipper effect” can propagate the molecule to more complete separation and form products such as those shown above. While not all molecules will dissociate in such a way, there is experimental evidence for this through significant concentrations of ethene and CO2 in the products.
Figure 7.
While initiation can occur at specific weak sites within the molecule, the “zipper effect” can propagate the molecule to more complete separation and form products such as those shown above. While not all molecules will dissociate in such a way, there is experimental evidence for this through significant concentrations of ethene and CO2 in the products.
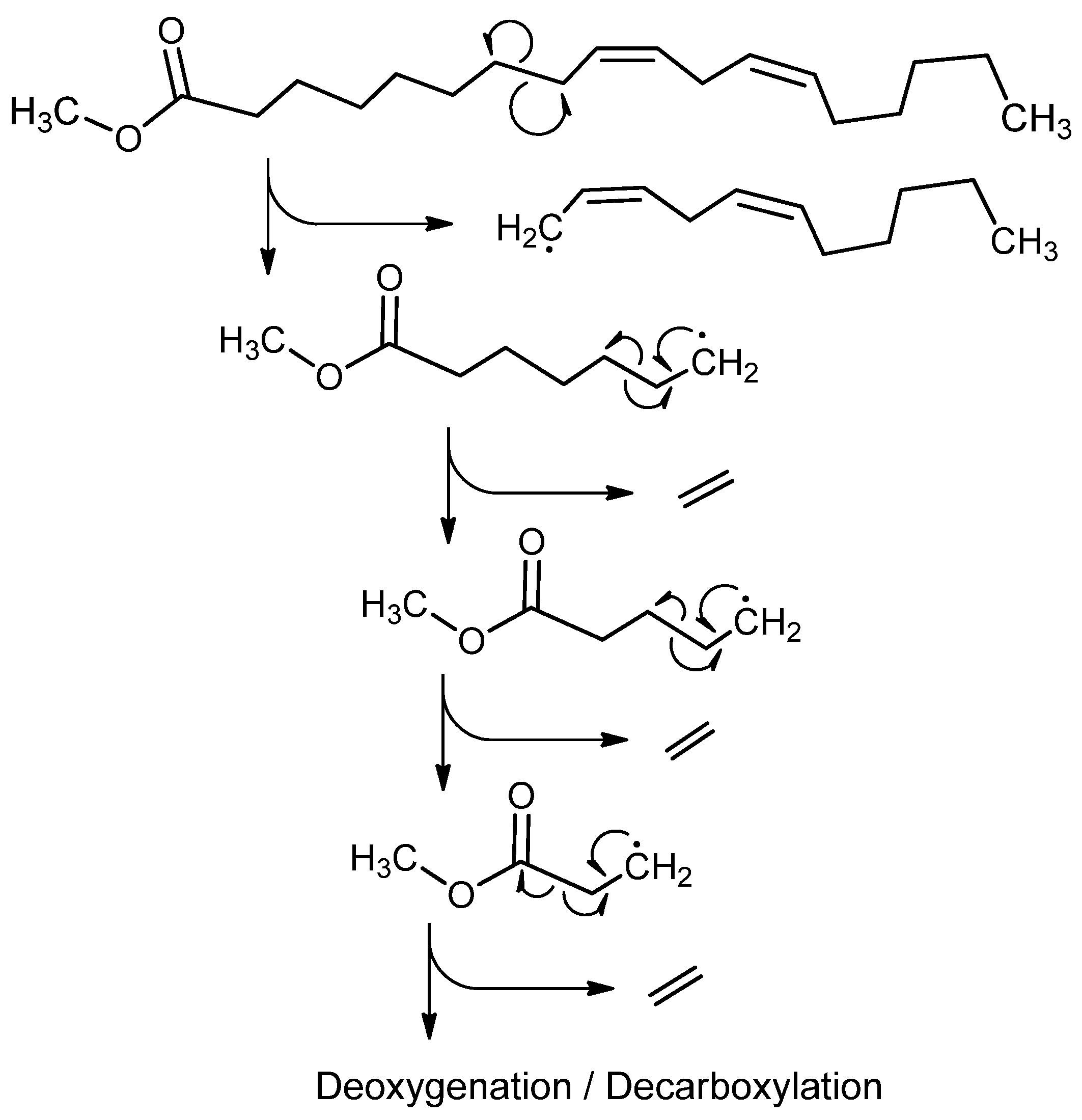
Figure 8.
Reaction pathways observed for deoxygenation/decarbonylation thermal deoxygenation. Reaction can occur as follows: decarbonylation (1), β-scission cascade (2), decarboxylation (3), pericyclic deoxygenation (4), methyl free-radical deoxygenation (5), α free-radical deoxygenation (6), or β free-radical deoxygenation (7).
Figure 8.
Reaction pathways observed for deoxygenation/decarbonylation thermal deoxygenation. Reaction can occur as follows: decarbonylation (1), β-scission cascade (2), decarboxylation (3), pericyclic deoxygenation (4), methyl free-radical deoxygenation (5), α free-radical deoxygenation (6), or β free-radical deoxygenation (7).
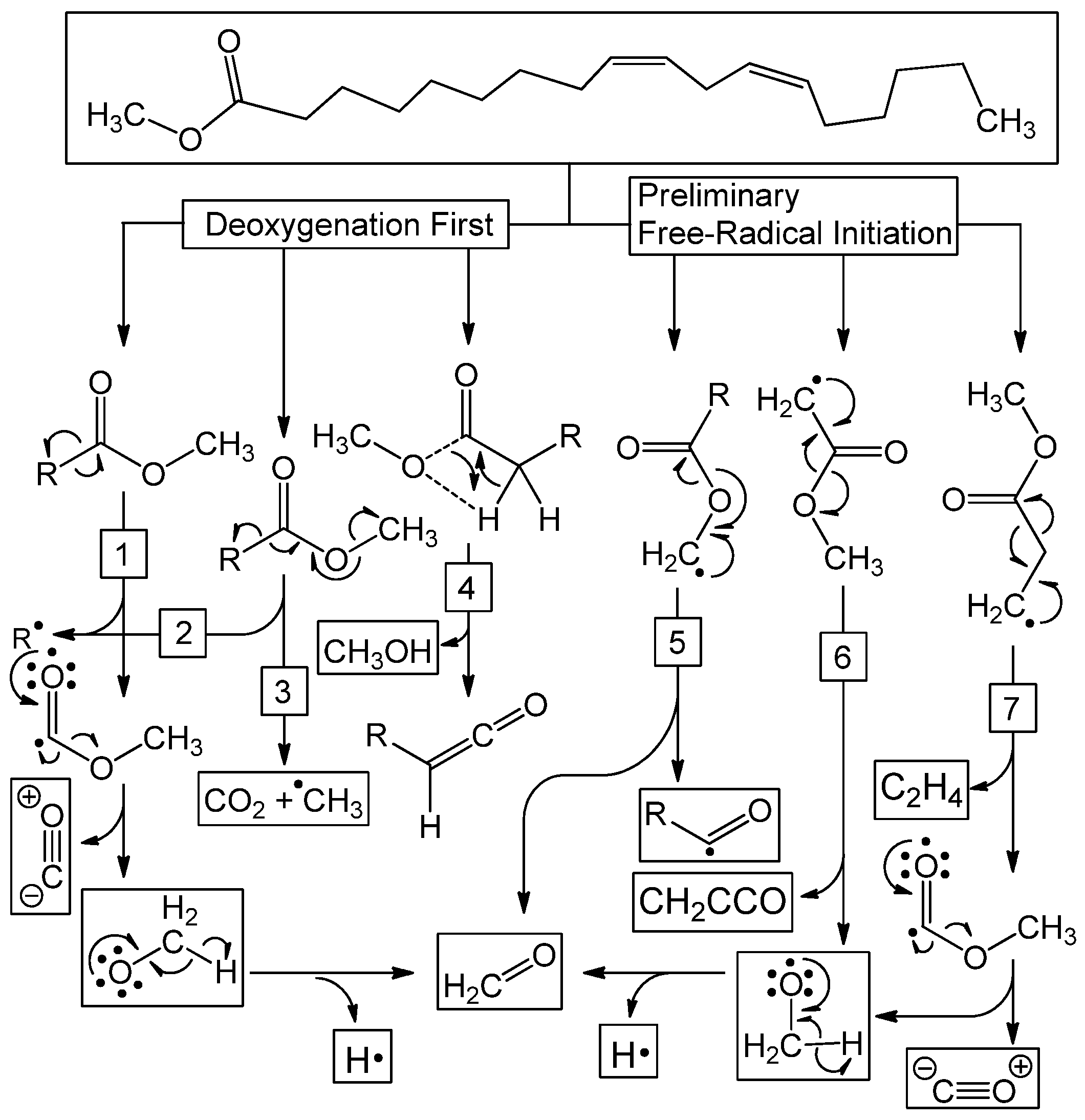
Figure 9.
Frequency of C–C bond dissociations grouped by pathways where it constitutes the first bond breakage and the cumulative observed frequency within the ensemble.
Figure 9.
Frequency of C–C bond dissociations grouped by pathways where it constitutes the first bond breakage and the cumulative observed frequency within the ensemble.

Figure 10.
Reaction scheme describing a probable complete dissociation of hydrocarbons. While this is a theoretical dissociation, each step was observed in a heightened frequency compared to abnormal dissociations.
Figure 10.
Reaction scheme describing a probable complete dissociation of hydrocarbons. While this is a theoretical dissociation, each step was observed in a heightened frequency compared to abnormal dissociations.

Figure 11.
Pericyclic reactions pertinent to the divinylcyclopropane molecule observed heavily in the experimental distribution. (a) Vinylcyclopropane rearrangement is a more common ring expansion reaction. (b) Divinylcyclopropane-cycloheptadiene rearrangement, this [3,3] sigmatropic shift derives much of its thermodynamic drive from the release of ring strain [53].
Figure 11.
Pericyclic reactions pertinent to the divinylcyclopropane molecule observed heavily in the experimental distribution. (a) Vinylcyclopropane rearrangement is a more common ring expansion reaction. (b) Divinylcyclopropane-cycloheptadiene rearrangement, this [3,3] sigmatropic shift derives much of its thermodynamic drive from the release of ring strain [53].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bakker, M.J.; Siebert, M.R. Atomistic Details of Methyl Linoleate Pyrolysis: Direct Molecular Dynamics Simulation of Converting Biodiesel to Petroleum Products. Energies 2024, 17, 2433. https://doi.org/10.3390/en17102433
AMA Style
Bakker MJ, Siebert MR. Atomistic Details of Methyl Linoleate Pyrolysis: Direct Molecular Dynamics Simulation of Converting Biodiesel to Petroleum Products. Energies. 2024; 17(10):2433. https://doi.org/10.3390/en17102433
Chicago/Turabian StyleBakker, Michael J., and Matthew R. Siebert. 2024. "Atomistic Details of Methyl Linoleate Pyrolysis: Direct Molecular Dynamics Simulation of Converting Biodiesel to Petroleum Products" Energies 17, no. 10: 2433. https://doi.org/10.3390/en17102433
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.