

Exogenous Metabolic Modulators Improve Response to Carboplatin in Triple-Negative Breast Cancer
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Generation of Inducible shATG5 Cell Lines
2.4. Western Immunoblotting
2.5. Autophagy Measurement
2.6. Viability Assays
2.7. Animal Study Design
3. Results
3.1. BMS-754807 Is Cytotoxic in TNBC Cells
3.2. Pretreatment with BMS-754807 Sensitizes TNBC Cells to CP
3.3. Pretreatment with HCQ Sensitizes TNBC Cells to CP
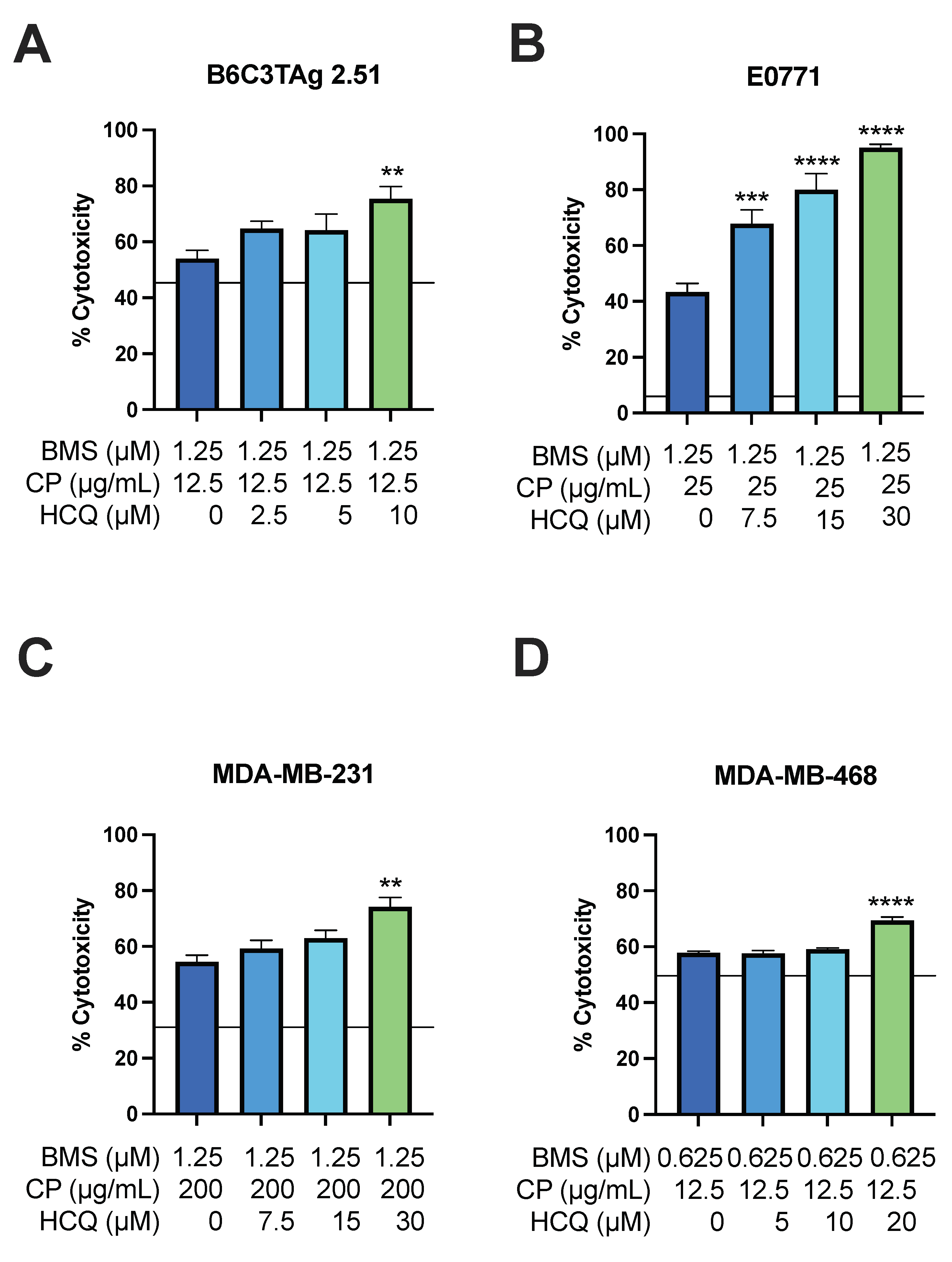
3.4. HCQ Treatment Sensitizes Viability of TNBC Cells to BMS-754807 and CP
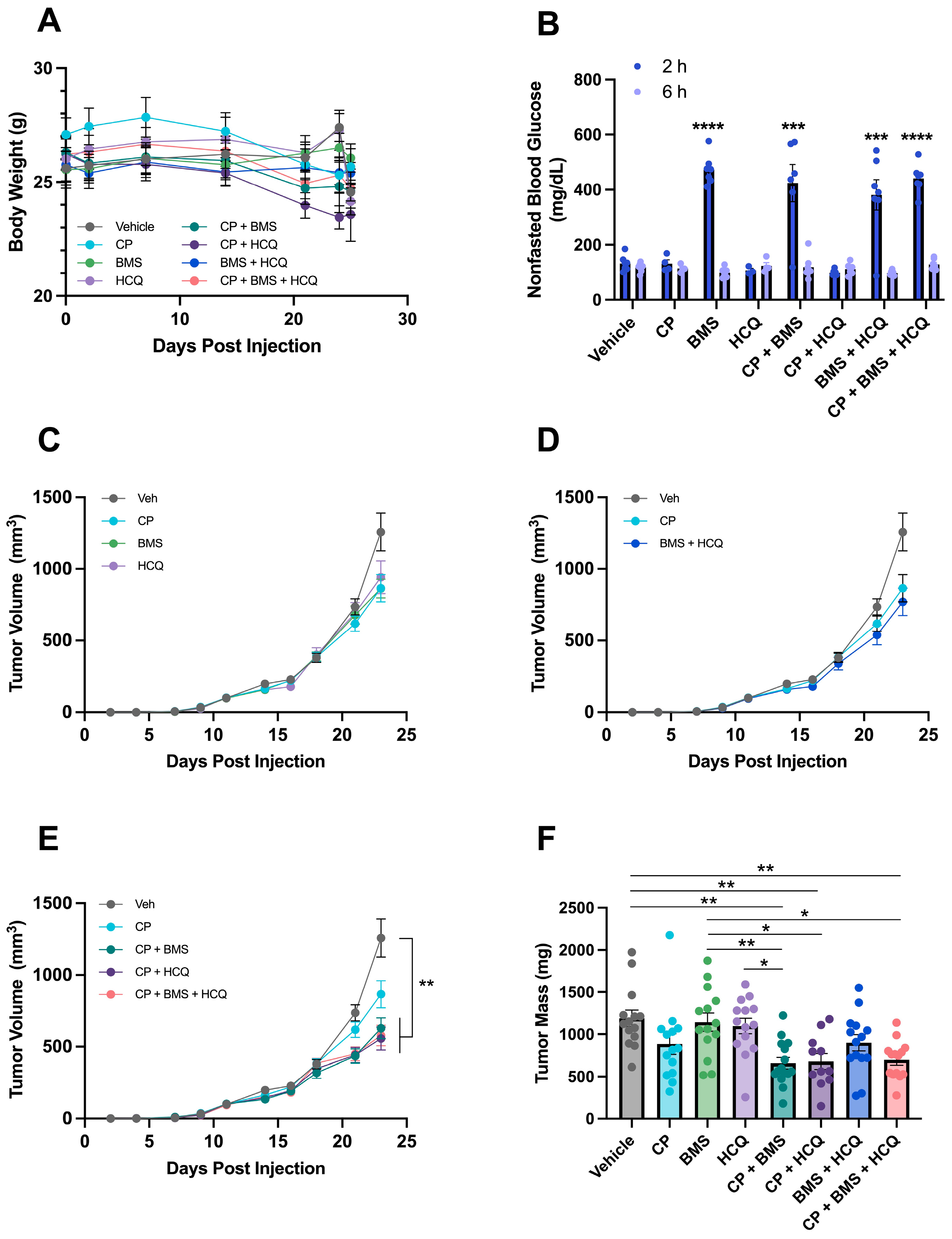
3.5. BMS-754807 and HCQ Sensitize metM-Wntlung Tumors to CP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Bonotto, M.; Gerratana, L.; Poletto, E.; Driol, P.; Giangreco, M.; Russo, S.; Minisini, A.M.; Andreetta, C.; Mansutti, M.; Pisa, F.E.; et al. Measures of outcome in metastatic breast cancer: Insights from a real-world scenario. Oncologist 2014, 19, 608–615. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Burstein, H.J.; Chew, H.; Dang, C.; et al. Breast Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 691–722. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Corti, C.; Schmid, P.; Cortes, J.; Mittendorf, E.A.; Rugo, H.; Tolaney, S.M.; Bianchini, G.; Andre, F.; Curigliano, G. Immunotherapy for early triple negative breast cancer: Research agenda for the next decade. npj Breast Cancer 2022, 8, 23. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Skinner, K.E.; Haiderali, A.; Huang, M.; Schwartzberg, L.S. Real-world effectiveness outcomes in patients diagnosed with metastatic triple-negative breast cancer. Future Oncol. 2021, 17, 931–941. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef]
- Martin, M.; Ramos-Medina, R.; Bernat, R.; Garcia-Saenz, J.A.; Del Monte-Millan, M.; Alvarez, E.; Cebollero, M.; Moreno, F.; Gonzalez-Haba, E.; Bueno, O.; et al. Activity of docetaxel, carboplatin, and doxorubicin in patient-derived triple-negative breast cancer xenografts. Sci. Rep. 2021, 11, 7064. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol. Lett. 2018, 15, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Yardley, D.A.; Layman, R.; Sparano, J.A.; Chuang, E.; Northfelt, D.W.; Schwartz, G.N.; Youssoufian, H.; Tang, S.; Novosiadly, R.; et al. Clinical and Translational Results of a Phase II, Randomized Trial of an Anti-IGF-1R (Cixutumumab) in Women with Breast Cancer That Progressed on Endocrine Therapy. Clin. Cancer Res. 2016, 22, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, M.; Holgersson, G.; Bondarenko, I.; Grechanaya, E.; Maximovich, A.; Andor, G.; Klockare, M.; Thureson, M.; Jerling, M.; Harmenberg, J. Phase II randomized study of the IGF-1R pathway modulator AXL1717 compared to docetaxel in patients with previously treated, locally advanced or metastatic non-small cell lung cancer. Acta Oncol. 2017, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; Rossi, E.L.; McDonell, S.B.; Chen, X.; Tsai, Y.H.; Parker, J.S.; Usary, J.; Perou, C.M.; Hursting, S.D. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 2017, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.S.; Glenny, E.M.; Kiesel, V.A.; Taylor, A.; Roth, D.; Albright, J.; VerHague, M.; French, J.E.; Coleman, M.F.; Hursting, S.D. Abstract 1336: Characterization of a novel transplantable model of obesity-driven basal-like breast cancer. Cancer Res. 2022, 82, 1336. [Google Scholar] [CrossRef]
- Oguri, S.; Sakakibara, T.; Mase, H.; Shimizu, T.; Ishikawa, K.; Kimura, K.; Smyth, R.D. Clinical Pharmacokinetics of Carboplatin. J. Clin. Pharmacol. 1988, 28, 208–215. [Google Scholar] [CrossRef]
- Stalnecker, C.A.; Grover, K.R.; Edwards, A.C.; Coleman, M.F.; Yang, R.; DeLiberty, J.M.; Papke, B.; Goodwin, C.M.; Pierobon, M.; Petricoin, E.F.; et al. Concurrent Inhibition of IGF1R and ERK Increases Pancreatic Cancer Sensitivity to Autophagy Inhibitors. Cancer Res. 2022, 82, 586–598. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S.; et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2017, 2, e89760. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, M.N.; Simeone, K.; Leclerc-Deslauniers, K.; Fleury, H.; Carmona, E.; Provencher, D.M.; Mes-Masson, A.M. Carboplatin response in preclinical models for ovarian cancer: Comparison of 2D monolayers, spheroids, ex vivo tumors and in vivo models. Sci. Rep. 2021, 11, 18183. [Google Scholar] [CrossRef]
- Van Eaton, K.M.; Gustafson, D.L. Pharmacokinetic and Pharmacodynamic Assessment of Hydroxychloroquine in Breast Cancer. J. Pharmacol. Exp. Ther. 2021, 379, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Moens, S.; Zhao, P.; Baietti, M.F.; Marinelli, O.; Van Haver, D.; Impens, F.; Floris, G.; Marangoni, E.; Neven, P.; Annibali, D.; et al. The mitotic checkpoint is a targetable vulnerability of carboplatin-resistant triple negative breast cancers. Sci. Rep. 2021, 11, 3176. [Google Scholar] [CrossRef] [PubMed]
- Hain, B.A.; Xu, H.; Wilcox, J.R.; Mutua, D.; Waning, D.L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- De Angel, R.E.; Blando, J.M.; Hogan, M.G.; Sandoval, M.A.; Lansakara, P.D.; Dunlap, S.M.; Hursting, S.D.; Cui, Z. Stearoyl gemcitabine nanoparticles overcome obesity-induced cancer cell resistance to gemcitabine in a mouse postmenopausal breast cancer model. Cancer Biol. Ther. 2013, 14, 357–364. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Philpott, N.J.; Elebute, M.O.; Powles, R.; Treleaven, J.G.; Gore, M.; Dainton, M.G.; Min, T.; Swansbury, G.J.; Catovsky, D. Platinum agents and secondary myeloid leukaemia: Two cases treated only with platinum-based drugs. Br. J. Haematol. 1996, 93, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Katagiri, H.; Katagiri, A.; Ishibashi, T.; Iida, K.; Nakayama, N.; Miyazaki, K. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following chemotherapy (paclitaxel and carboplatin) and radiation therapy in ovarian cancer: A case report. Eur. J. Gynaecol. Oncol. 2014, 35, 443–448. [Google Scholar] [PubMed]
- Yusoh, N.A.; Ahmad, H.; Gill, M.R. Combining PARP Inhibition with Platinum, Ruthenium or Gold Complexes for Cancer Therapy. ChemMedChem 2020, 15, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Ewens, A.; Mihich, E.; Ehrke, M.J. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer. Res. 2005, 25, 3905–3915. [Google Scholar]
- Jenkins, D.E.; Hornig, Y.S.; Oei, Y.; Dusich, J.; Purchio, T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005, 7, R444–R454. [Google Scholar] [CrossRef] [PubMed]
- Vantyghem, S.A.; Allan, A.L.; Postenka, C.O.; Al-Katib, W.; Keeney, M.; Tuck, A.B.; Chambers, A.F. A new model for lymphatic metastasis: Development of a variant of the MDA-MB-468 human breast cancer cell line that aggressively metastasizes to lymph nodes. Clin. Exp. Metastasis 2005, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.E.; Jones, R.A.; Briah, R.; Murray, P.; Moorehead, R.A. BMS-754807 is cytotoxic to non-small cell lung cancer cells and enhances the effects of platinum chemotherapeutics in the human lung cancer cell line A549. BMC Res. Notes 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Zhu, J.; Zheng, H.; Chen, S.; Chen, L.; Yang, H.S. IGF-1R inhibition induces MEK phosphorylation to promote survival in colon carcinomas. Signal Transduct. Target. Ther. 2020, 5, 153. [Google Scholar] [CrossRef]
- Eckstein, N.; Servan, K.; Hildebrandt, B.; Politz, A.; von Jonquieres, G.; Wolf-Kummeth, S.; Napierski, I.; Hamacher, A.; Kassack, M.U.; Budczies, J.; et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009, 69, 2996–3003. [Google Scholar] [CrossRef]
- Oza, A.; Kaye, S.; Van Tornout, J.; Sessa, C.; Gore, M.; Naumann, R.W.; Hirte, H.; Colombo, N.; Chen, J.; Gorla, S.; et al. Phase 2 study evaluating intermittent and continuous linsitinib and weekly paclitaxel in patients with recurrent platinum resistant ovarian epithelial cancer. Gynecol. Oncol. 2018, 149, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yin, X.; Languino, L.R.; Altieri, D.C. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat. Biopharm. Res. 2018, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; O’Shea, S.; Lyons, A.; Fogarty, F.M.; McCabe, N.; Kennedy, R.D.; O’Connor, R. IGF-1R inhibition sensitizes breast cancer cells to ATM-related kinase (ATR) inhibitor and cisplatin. Oncotarget 2016, 7, 56826–56841. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.E.; Lashinger, L.M.; Otto, G.; Nunez, N.P.; Hursting, S.D. Decreased systemic IGF-1 in response to calorie restriction modulates murine tumor cell growth, nuclear factor-kappaB activation, and inflammation-related gene expression. Mol. Carcinog. 2013, 52, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- de Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W.; et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef] [PubMed]
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- Bernard, M.; Cardin, G.B.; Cahuzac, M.; Ayad, T.; Bissada, E.; Guertin, L.; Bahig, H.; Nguyen-Tan, P.F.; Filion, E.; Ballivy, O.; et al. Dual Inhibition of Autophagy and PI3K/AKT/MTOR Pathway as a Therapeutic Strategy in Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2371. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | BMS-754807 (µM) | CP (µg/mL) | Synergy Score | Cytotoxicity (%) | Highest Synergy Score |
|---|---|---|---|---|---|
| E0771 | 1.25 | 25 | 12.74 | 47.9 | 15.77 |
| B6C3TAg 2.51 | 1.25 | 12.5 | 34.71 | 56.0 | 34.71 |
| metM-Wntlung | 1.25 | 25 | 14.19 | 58.8 | 14.50 |
| MDA-MB-231 | 1.25 | 200 | 13.98 | 56.8 | 13.98 |
| MDA-MB-468 | 0.625 | 12.5 | 14.87 | 53.5 | 14.87 |
| Cell Line | HCQ (µM) | CP (µg/mL) | Highest Synergy Score | Cytotoxicity (%) |
|---|---|---|---|---|
| E0771 | 15 | 50 | 2.67 | 91.3 |
| B6C3TAg 2.51 | 2.5 | 25 | 0.67 | 85.0 |
| metM-Wntlung | 5 | 25 | 8.71 | 69.7 |
| MDA-MB-231 | 5 | 200 | 12.42 | 41.1 |
| MDA-MB-468 | 3.75 | 12.5 | 15.07 | 60.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, A.N.; Kiesel, V.A.; Gates, C.E.; Brosnan, B.H.; Connelly, S.P.; Glenny, E.M.; Cozzo, A.J.; Hursting, S.D.; Coleman, M.F. Exogenous Metabolic Modulators Improve Response to Carboplatin in Triple-Negative Breast Cancer. Cells 2024, 13, 806. https://doi.org/10.3390/cells13100806
Ho AN, Kiesel VA, Gates CE, Brosnan BH, Connelly SP, Glenny EM, Cozzo AJ, Hursting SD, Coleman MF. Exogenous Metabolic Modulators Improve Response to Carboplatin in Triple-Negative Breast Cancer. Cells. 2024; 13(10):806. https://doi.org/10.3390/cells13100806
Chicago/Turabian StyleHo, Alyssa N., Violet A. Kiesel, Claire E. Gates, Bennett H. Brosnan, Scott P. Connelly, Elaine M. Glenny, Alyssa J. Cozzo, Stephen D. Hursting, and Michael Francis Coleman. 2024. "Exogenous Metabolic Modulators Improve Response to Carboplatin in Triple-Negative Breast Cancer" Cells 13, no. 10: 806. https://doi.org/10.3390/cells13100806