
Molecular Regulation of Fetal Brain Development in Inbred and Congenic Mouse Strains Differing in Longevity



Abstract
1. Introduction
2. Materials and Methods
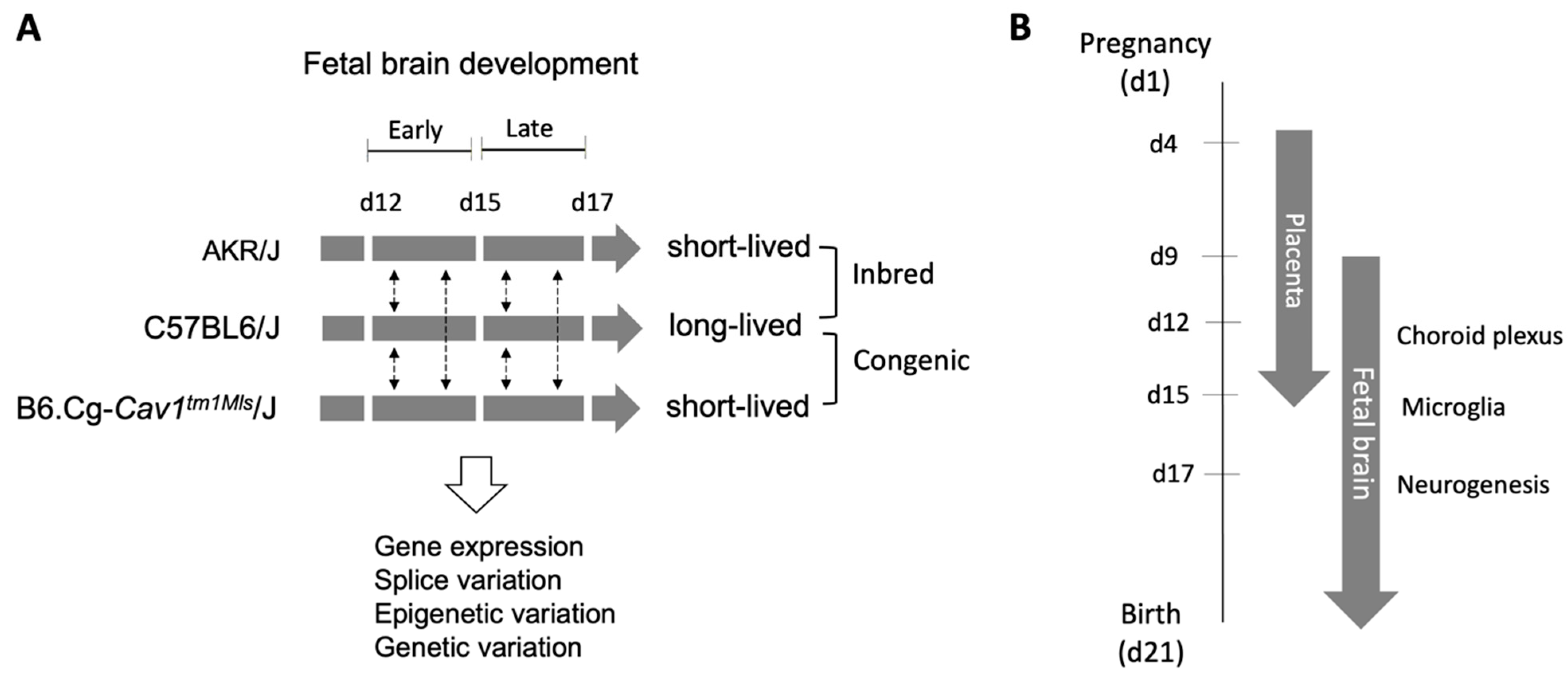
2.1. Overview of Approach
2.2. Animals and Sample Collection
2.3. RNA Sequencing
2.4. RNA-Seq Data Analysis
2.5. Detection of Novel and Known Splice Variants in Developing Fetal Brain
2.6. Analysis of Epigenetic Variation
2.7. Genetic Variant Analysis
3. Results
3.1. Gene Expression Variation in the Fetal Brain of Short- and Long-Lived Mice
3.2. Functional Annotation of the Differentially Expressed Genes in the Fetal Brain
3.3. Alternative Splice Variants in the Fetal Brain of Short- versus Long-Lived Mice
3.4. DNA Methylation Changes in the Fetal Brain between Strains
3.5. Influence of Genetic Variation on the Regulation of Fetal Brain Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dalgaard, C.-J.; Hansen, C.W.; Strulik, H. Fetal Origins-A Life Cycle Model of Health and Aging from Conception to Death. Health Econ. 2021, 30, 1276–1290. [Google Scholar] [CrossRef]
- Hadad, N.; Masser, D.R.; Blanco-Berdugo, L.; Stanford, D.R.; Freeman, W.M. Early-Life DNA Methylation Profiles Are Indicative of Age-Related Transcriptome Changes. Epigenetics Chromatin 2019, 12, 58. [Google Scholar] [CrossRef]
- Tamnes, C.K.; Walhovd, K.B.; Dale, A.M.; Østby, Y.; Grydeland, H.; Richardson, G.; Westlye, L.T.; Roddey, J.C.; Hagler, D.J.; Due-Tønnessen, P.; et al. Brain Development and Aging: Overlapping and Unique Patterns of Change. Neuroimage 2013, 68, 63–74. [Google Scholar] [CrossRef]
- Early Life Origins of Ageing and Longevity; Vaiserman, A., Ed.; Healthy Ageing and Longevity; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-030-24957-1. [Google Scholar]
- Marchionni, S.; Sell, C.; Lorenzini, A. Development and Longevity: Cellular and Molecular Determinants—A Mini-Review. Gerontology 2020, 66, 223–230. [Google Scholar] [CrossRef]
- Dilman, V.M. Age-Associated Elevation of Hypothalamic, Threshold to Feedback Control, and Its Role in Development, Ageine, and Disease. Lancet 1971, 1, 1211–1219. [Google Scholar] [CrossRef]
- Feltes, B.C.; de Faria Poloni, J.; Bonatto, D. Development and Aging: Two Opposite but Complementary Phenomena. Interdiscip. Top. Gerontol. 2015, 40, 74–84. [Google Scholar] [CrossRef]
- de Magalhães, J.P.; Church, G.M. Genomes Optimize Reproduction: Aging as a Consequence of the Developmental Program. Physiology 2005, 20, 252–259. [Google Scholar] [CrossRef]
- Bartke, A. Early Life Events Can Shape Aging and Longevity. Curr. Aging Sci. 2015, 8, 11–13. [Google Scholar] [CrossRef] [PubMed]
- MacInnes, A.W. The Role of the Ribosome in the Regulation of Longevity and Lifespan Extension. Wiley Interdiscip. Rev. RNA 2016, 7, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Pitale, S.; Sahasrabuddhe, A. Fetal Origin of Vascular Aging. Indian. J. Endocrinol. Metab. 2011, 15 (Suppl. 4), S289–S297. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, C.L. Nutritional Factors in Fetal and Infant Brain Development. Ann. Nutr. Metab. 2019, 75 (Suppl. 1), 20–32. [Google Scholar] [CrossRef] [PubMed]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75 (Suppl. 1), 8–18. [Google Scholar] [CrossRef]
- Aguilaniu, H. The Mysterious Relationship between Reproduction and Longevity. Worm 2015, 4, e1020276. [Google Scholar] [CrossRef]
- Antebi, A. Regulation of Longevity by the Reproductive System. Exp. Gerontol. 2013, 48, 596–602. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Thornburg, K.L.; Kajantie, E.; Eriksson, J.G. The Lifespan of Men and the Shape of Their Placental Surface at Birth. Placenta 2011, 32, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Templeman, N.M.; Murphy, C.T. Regulation of Reproduction and Longevity by Nutrient-Sensing Pathways. J. Cell Biol. 2018, 217, 93–106. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Scheibye-Knudsen, M.; Longo, D.L.; de Cabo, R. Animal Models of Aging Research: Implications for Human Aging and Age-Related Diseases. Annu. Rev. Anim. Biosci. 2015, 3, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.L.H.; Roth, T.L. Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior. Genes 2019, 10, 47. [Google Scholar] [CrossRef]
- Herr, W.; Gilbert, W. Germ-Line MuLV Reintegrations in AKR/J Mice. Nature 1982, 296, 865–868. [Google Scholar] [CrossRef]
- Hays, E.F.; Beck, W.S. The Development of Leukemia and Other Neoplasms in Mice Receiving Cell-Free Extracts from a High-Leukemia (AKR) Strain. Cancer Res. 1958, 18, 676–681. [Google Scholar]
- Langlois, M.-A.; Kemmerich, K.; Rada, C.; Neuberger, M.S. The AKV Murine Leukemia Virus Is Restricted and Hypermutated by Mouse APOBEC3. J. Virol. 2009, 83, 11550–11559. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Tsaih, S.-W.; Petkova, S.B.; de Evsikova, C.M.; Xing, S.; Marion, M.A.; Bogue, M.A.; Mills, K.D.; Peters, L.L.; Bult, C.J.; et al. Aging in Inbred Strains of Mice: Study Design and Interim Report on Median Lifespans and Circulating IGF1 Levels. Aging Cell 2009, 8, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Velasco, G.; López-Otín, C. Mouse Models to Disentangle the Hallmarks of Human Aging. Circ. Res. 2018, 123, 905–924. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-H.; Feng, D.; Nagy, J.A.; Sciuto, T.E.; Dvorak, A.M.; Dvorak, H.F. Vascular Permeability and Pathological Angiogenesis in Caveolin-1-Null Mice. Am. J. Pathol. 2009, 175, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Woodman, S.E.; Park, D.S.; Lisanti, M.P. Caveolin, Caveolae, and Endothelial Cell Function. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Behura, S.K. Role of Caveolin-1 in Metabolic Programming of Fetal Brain. iScience 2023, 26, 107710. [Google Scholar] [CrossRef]
- Park, D.S.; Cohen, A.W.; Frank, P.G.; Razani, B.; Lee, H.; Williams, T.M.; Chandra, M.; Shirani, J.; De Souza, A.P.; Tang, B.; et al. Caveolin-1 Null (−/−) Mice Show Dramatic Reductions in Life Span. Biochemistry 2003, 42, 15124–15131. [Google Scholar] [CrossRef]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 Null Mice Are Viable but Show Evidence of Hyperproliferative and Vascular Abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of Caveolin-1 Accelerates Neurodegeneration and Aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef]
- Hattori, C.; Asai, M.; Onishi, H.; Sasagawa, N.; Hashimoto, Y.; Saido, T.C.; Maruyama, K.; Mizutani, S.; Ishiura, S. BACE1 Interacts with Lipid Raft Proteins. J. Neurosci. Res. 2006, 84, 912–917. [Google Scholar] [CrossRef]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, Cellular Senescence and Age-Related Diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.E.; Copp, A.J. Neural Tube Defects. Annu. Rev. Neurosci. 2014, 37, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.S.; Morrison, J.P.; Southwell, M.F.; Foley, J.F.; Bolon, B.; Elmore, S.A. Histology Atlas of the Developing Prenatal and Postnatal Mouse Central Nervous System, with Emphasis on Prenatal Days E7.5 to E18.5. Toxicol. Pathol. 2017, 45, 705–744. [Google Scholar] [CrossRef] [PubMed]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef]
- Behura, S.K.; Dhakal, P.; Kelleher, A.M.; Balboula, A.; Patterson, A.; Spencer, T.E. The Brain-Placental Axis: Therapeutic and Pharmacological Relevancy to Pregnancy. Pharmacol. Res. 2019, 149, 104468. [Google Scholar] [CrossRef]
- Broad, K.D.; Keverne, E.B. Placental Protection of the Fetal Brain during Short-Term Food Deprivation. Proc. Natl. Acad. Sci. USA 2011, 108, 15237–15241. [Google Scholar] [CrossRef]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth—New Insights From Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef]
- Watson, E.D.; Cross, J.C. Development of Structures and Transport Functions in the Mouse Placenta. Physiology 2005, 20, 180–193. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Samal, A.; Davis, D.J.; Behura, S.K. Ablation of Placental REST Deregulates Fetal Brain Metabolism and Impacts Gene Expression of the Offspring Brain at the Postnatal and Adult Stages. FASEB J. 2024, 38, e23349. [Google Scholar] [CrossRef]
- Islam, M.; Strawn, M.; Behura, S.K. Fetal Origin of Sex-Bias Brain Aging. FASEB J. 2022, 36, e22463. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.-K.; Krueger, F.; BI Ageing Clock Team; von Meyenn, F.; Stegle, O.; Reik, W. Multi-Tissue DNA Methylation Age Predictor in Mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.; Mitrea, C.; Nguyen, T.; Draghici, S. A Survey of the Approaches for Identifying Differential Methylation Using Bisulfite Sequencing Data. Brief. Bioinform. 2017, 19, 737–753. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R Package for Visualizing, Adjusting and Comparing Trees of Hierarchical Clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen Diverse Laboratory Mouse Reference Genomes Define Strain-Specific Haplotypes and Novel Functional Loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef]
- McHugh, M.L. The Chi-Square Test of Independence. Biochem. Med. 2013, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-Scale Gene Function Analysis with the PANTHER Classification System. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; Dhananjaya, D.; Tarn, W.-Y. Alternative Splicing in Neurogenesis and Brain Development. Front. Mol. Biosci. 2018, 5, 12. [Google Scholar] [CrossRef]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of Canonical and Non-Canonical Splice Sites in Mammalian Genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef]
- Meer, M.V.; Podolskiy, D.I.; Tyshkovskiy, A.; Gladyshev, V.N. A Whole Lifespan Mouse Multi-Tissue DNA Methylation Clock. Elife 2018, 7, e40675. [Google Scholar] [CrossRef]
- Kozak, C.A.; Rowe, W.P. Genetic Mapping of the Ecotropic Virus-Inducing Locus Akv-2 of the AKR Mouse. J. Exp. Med. 1980, 152, 1419–1423. [Google Scholar] [CrossRef]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B. Alternative Splicing in Aging and Longevity. Hum. Genet. 2020, 139, 357–369. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of Alternative Splicing and Its Regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Flint, M.S.; Bovbjerg, D.H. DNA Damage as a Result of Psychological Stress: Implications for Breast Cancer. Breast Cancer Res. 2012, 14, 320. [Google Scholar] [CrossRef]
- Meyer, F.; Engel, A.M.; Krause, A.K.; Wagner, T.; Poole, L.; Dubrovska, A.; Peitzsch, C.; Rothkamm, K.; Petersen, C.; Borgmann, K. Efficient DNA Repair Mitigates Replication Stress Resulting in Less Immunogenic Cytosolic DNA in Radioresistant Breast Cancer Stem Cells. Front. Immunol. 2022, 13, 765284. [Google Scholar] [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The Central Role of DNA Damage in the Ageing Process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA Methylation and Gene Expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Bacon, E.R.; Brinton, R.D. Epigenetics of the Developing and Aging Brain: Mechanisms That Regulate Onset and Outcomes of Brain Reorganization. Neurosci. Biobehav. Rev. 2021, 125, 503–516. [Google Scholar] [CrossRef]
- Numata, S.; Ye, T.; Hyde, T.M.; Guitart-Navarro, X.; Tao, R.; Wininger, M.; Colantuoni, C.; Weinberger, D.R.; Kleinman, J.E.; Lipska, B.K. DNA Methylation Signatures in Development and Aging of the Human Prefrontal Cortex. Am. J. Hum. Genet. 2012, 90, 260–272. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Raichle, M.E.; Gusnard, D.A. Appraising the Brain’s Energy Budget. PNAS 2002, 99, 10237–10239. [Google Scholar] [CrossRef]
- Lust, W.D.; Pundik, S.; Zechel, J.; Zhou, Y.; Buczek, M.; Selman, W.R. Changing Metabolic and Energy Profiles in Fetal, Neonatal, and Adult Rat Brain. Metab. Brain Dis. 2003, 18, 195–206. [Google Scholar] [CrossRef]
- Rogne, T.; Engstrøm, A.A.; Jacobsen, G.W.; Skranes, J.; Østgård, H.F.; Martinussen, M. Fetal Growth, Cognitive Function, and Brain Volumes in Childhood and Adolescence. Obstet. Gynecol. 2015, 125, 673–682. [Google Scholar] [CrossRef]
- Bonnin, A.; Goeden, N.; Chen, K.; Wilson, M.L.; King, J.; Shih, J.C.; Blakely, R.D.; Deneris, E.S.; Levitt, P. A Transient Placental Source of Serotonin for the Fetal Forebrain. Nature 2011, 472, 347–350. [Google Scholar] [CrossRef]
- Bronson, S.L.; Bale, T.L. The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacology 2016, 41, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Placental Serotonin Signaling, Pregnancy Outcomes, and Regulation of Fetal Brain Development†. Biol. Reprod. 2020, 102, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Zeltser, L.M.; Leibel, R.L. Roles of the Placenta in Fetal Brain Development. Proc. Natl. Acad. Sci. USA 2011, 108, 15667–15668. [Google Scholar] [CrossRef]
- Barton, R.A.; Capellini, I. Maternal Investment, Life Histories, and the Costs of Brain Growth in Mammals. Proc. Natl. Acad. Sci. USA 2011, 108, 6169–6174. [Google Scholar] [CrossRef]
- Lindsay, K.L.; Buss, C.; Wadhwa, P.D.; Entringer, S. The Interplay Between Nutrition and Stress in Pregnancy: Implications for Fetal Programming of Brain Development. Biol. Psychiatry 2019, 85, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, D.A.; Hutton, O.; Hopkins, R.; Cagampang, F.; Smyth, N.R.; Fleming, T.P.; Eckert, J.; Willaime-Morawek, S. Preimplantation or Gestation/Lactation High-Fat Diet Alters Adult Offspring Metabolism and Neurogenesis. Brain Commun. 2023, 5, fcad093. [Google Scholar] [CrossRef]
- Singh, S.K.; Kagalwala, M.N.; Parker-Thornburg, J.; Adams, H.; Majumder, S. REST Maintains Self-Renewal and Pluripotency of Embryonic Stem Cells. Nature 2008, 453, 223–227. [Google Scholar] [CrossRef]
- Zullo, J.M.; Drake, D.; Aron, L.; O’Hern, P.; Dhamne, S.C.; Davidsohn, N.; Mao, C.-A.; Klein, W.H.; Rotenberg, A.; Bennett, D.A.; et al. Regulation of Lifespan by Neural Excitation and REST. Nature 2019, 574, 359–364. [Google Scholar] [CrossRef]
- Lind, M.I.; Spagopoulou, F. Evolutionary Consequences of Epigenetic Inheritance. Heredity 2018, 121, 205–209. [Google Scholar] [CrossRef]
- Hou, C. The Energy Trade-off between Growth and Longevity. Mech. Ageing Dev. 2013, 134, 373–380. [Google Scholar] [CrossRef]
- Park, S.E.; Kim, H.S.; Kwon, S.J.; Kim, M.-J.; Choi, S.; Oh, S.; Ryu, G.H.; Jeon, H.B.; Na, D.L.; Chang, J.W. Exposure of Mesenchymal Stem Cells to an Alzheimer’s Disease Environment Enhances Therapeutic Effects. Stem Cells Int. 2021, 2021, e6660186. [Google Scholar] [CrossRef]
- Srivastava, R.; Li, A.; Datta, T.; Jha, N.K.; Talukder, S.; Jha, S.K.; Chen, Z.-S. Advances in Stromal Cell Therapy for Management of Alzheimer’s Disease. Front. Pharmacol. 2022, 13, 955401. [Google Scholar] [CrossRef]
- Regmi, S.; Liu, D.D.; Shen, M.; Kevadiya, B.D.; Ganguly, A.; Primavera, R.; Chetty, S.; Yarani, R.; Thakor, A.S. Mesenchymal Stromal Cells for the Treatment of Alzheimer’s Disease: Strategies and Limitations. Front. Mol. Neurosci. 2022, 15, 1011225. [Google Scholar] [CrossRef]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Meyers, C.A.; Albitar, M.; Estey, E. Cognitive Impairment, Fatigue, and Cytokine Levels in Patients with Acute Myelogenous Leukemia or Myelodysplastic Syndrome. Cancer 2005, 104, 788–793. [Google Scholar] [CrossRef]
- Williams, A.M.; van Wijngaarden, E.; Seplaki, C.L.; Heckler, C.; Weber, M.T.; Barr, P.M.; Zent, C.S.; Janelsins, M.C. Cognitive Function in Patients with Chronic Lymphocytic Leukemia: A Cross-Sectional Study Examining Effects of Disease and Treatment. Leuk. Lymphoma 2020, 61, 1627–1635. [Google Scholar] [CrossRef]
- Tylee, D.S.; Kawaguchi, D.M.; Glatt, S.J. On the Outside, Looking in: A Review and Evaluation of the Comparability of Blood and Brain “-Omes”. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 595–603. [Google Scholar] [CrossRef]
- Walton, E.; Hass, J.; Liu, J.; Roffman, J.L.; Bernardoni, F.; Roessner, V.; Kirsch, M.; Schackert, G.; Calhoun, V.; Ehrlich, S. Correspondence of DNA Methylation Between Blood and Brain Tissue and Its Application to Schizophrenia Research. Schizophr. Bull. 2016, 42, 406–414. [Google Scholar] [CrossRef]
- Edgar, R.D.; Jones, M.J.; Meaney, M.J.; Turecki, G.; Kobor, M.S. BECon: A Tool for Interpreting DNA Methylation Findings from Blood in the Context of Brain. Transl. Psychiatry 2017, 7, e1187. [Google Scholar] [CrossRef]
- Farré, P.; Jones, M.J.; Meaney, M.J.; Emberly, E.; Turecki, G.; Kobor, M.S. Concordant and Discordant DNA Methylation Signatures of Aging in Human Blood and Brain. Epigenetics Chromatin 2015, 8, 19. [Google Scholar] [CrossRef]
- Lin, D.; Chen, J.; Ehrlich, S.; Bustillo, J.R.; Perrone-Bizzozero, N.; Walton, E.; Clark, V.P.; Wang, Y.-P.; Sui, J.; Du, Y.; et al. Cross-Tissue Exploration of Genetic and Epigenetic Effects on Brain Gray Matter in Schizophrenia. Schizophr. Bull. 2018, 44, 443–452. [Google Scholar] [CrossRef]
- Strawn, M.; Safranski, T.J.; Behura, S.K. Does DNA Methylation in the Fetal Brain Leave an Epigenetic Memory in the Blood? Gene 2023, 887, 147788. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Wang, Z.; Hortobágyi, T.; Witten, J.T.; Zarnack, K.; Kayikci, M.; Clark, T.A.; Schweitzer, A.C.; Rot, G.; Curk, T.; et al. Analysis of Alternative Splicing Associated with Aging and Neurodegeneration in the Human Brain. Genome Res. 2011, 21, 1572–1582. [Google Scholar] [CrossRef]
- Takata, A.; Matsumoto, N.; Kato, T. Genome-Wide Identification of Splicing QTLs in the Human Brain and Their Enrichment among Schizophrenia-Associated Loci. Nat. Commun. 2017, 8, 14519. [Google Scholar] [CrossRef]
- Mazin, P.; Xiong, J.; Liu, X.; Yan, Z.; Zhang, X.; Li, M.; He, L.; Somel, M.; Yuan, Y.; Phoebe Chen, Y.-P.; et al. Widespread Splicing Changes in Human Brain Development and Aging. Mol. Syst. Biol. 2013, 9, 633. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP-DM Distance Bins | SNP Count (Within Bin) | DM Count (Within Bin) | SNP Count (Not in the Bin) | DM Count (Not in the Bin) | Odds Ratio | p-Value |
|---|---|---|---|---|---|---|
| 0–0.1 kb | 38 | 170 | 277 | 1787 | 1.4418 | 0.03737 |
| 0.1–1 kb | 62 | 302 | 254 | 1655 | 1.3375 | 0.03802 |
| 1–5 kb | 55 | 357 | 252 | 1600 | 0.9782 | 0.58150 |
| 5–10 kb | 23 | 162 | 282 | 1795 | 0.9038 | 0.70291 |
| 10–50 kb | 87 | 609 | 217 | 1348 | 0.8875 | 0.82764 |
| 50–100 kb | 27 | 177 | 276 | 1780 | 0.9838 | 0.56439 |
| 100–200 kb | 20 | 144 | 283 | 1813 | 0.8898 | 0.71749 |
| 200–400 kb | 4 | 20 | 299 | 1937 | 1.2955 | 0.40478 |
| 400–800 kb | 2 | 11 | 301 | 1946 | 1.1754 | 0.53688 |
| 800–1000 kb | 1 | 5 | 302 | 1952 | 1.2926 | 0.57884 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.; Behura, S.K. Molecular Regulation of Fetal Brain Development in Inbred and Congenic Mouse Strains Differing in Longevity. Genes 2024, 15, 604. https://doi.org/10.3390/genes15050604
Islam M, Behura SK. Molecular Regulation of Fetal Brain Development in Inbred and Congenic Mouse Strains Differing in Longevity. Genes. 2024; 15(5):604. https://doi.org/10.3390/genes15050604
Chicago/Turabian StyleIslam, Maliha, and Susanta K. Behura. 2024. "Molecular Regulation of Fetal Brain Development in Inbred and Congenic Mouse Strains Differing in Longevity" Genes 15, no. 5: 604. https://doi.org/10.3390/genes15050604
APA StyleIslam, M., & Behura, S. K. (2024). Molecular Regulation of Fetal Brain Development in Inbred and Congenic Mouse Strains Differing in Longevity. Genes, 15(5), 604. https://doi.org/10.3390/genes15050604