
Nanopore-Based Surveillance of Leishmania Parasites in Culicoides Latrielle (Diptera: Ceratopogonidae) Caught from the Affected Community and Tham Phra Cave in Chiang Rai Province, the Endemic Area of Leishmaniasis in Northern Thailand
 , , , , , and
, , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
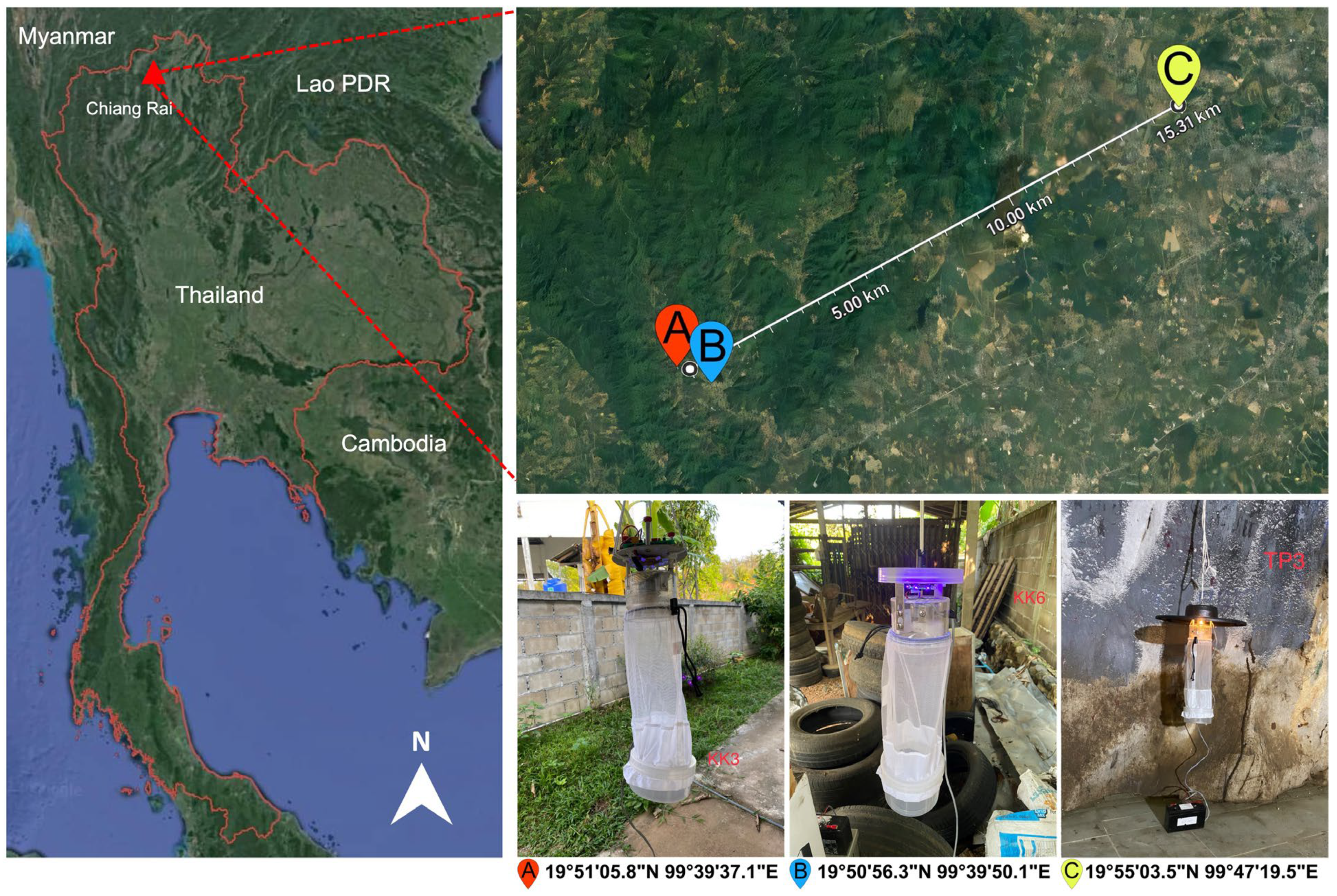
2.2. Investigation Areas, Biting Midge Collection, and Morphological Identification
2.3. Genomic DNA Isolation from Culicoides Samples
2.4. Molecular Screening of Leishmania DNA by ITS1-PCR
2.5. MinION® Amplicon Sequencing and ITS1 Consensus Calling
2.6. BLASTn Alignment and Phylogenetic Analysis of ITS1 Consensus Sequences
2.7. Haplotype Network Analysis
3. Results
3.1. Species Composition and Relative Abundance of Culicoides Species
3.2. Prevalence of Leishmania Infection in Culicoides and ITS1 Amplicon Sequencing
3.3. Molecular Identification and Phylogenetic Confirmation of Leishmania Species
3.4. Genetic Diversity and Neutrality Test of Leishmania ITS1 Haplotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 26 April 2024).
- Espinosa, O.A.; Serrano, M.G.; Camargo, E.P.; Teixeira, M.M.G.; Shaw, J.J. An appraisal of the taxonomy and nomenclature of trypanosomatids presently classified as Leishmania and Endotrypanum. Parasitology 2016, 145, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Muniz, J.; Medina, H. Cutaneous leishmaniasis of the guinea pig, Leishmania enriettii n. sp. Hospital 1948, 33, 7–25. [Google Scholar] [PubMed]
- Pothirat, T.; Tantiworawit, A.; Chaiwarith, R.; Jariyapan, N.; Wannasan, A.; Siriyasatien, P.; Supparatpinyo, K.; Bates, M.D.; Kwakye-Nuako, G.; Bates, P. First isolation of Leishmania from Northern Thailand: Case report, identification as Leishmania martiniquensis and Phylogenetic Position within the Leishmania enriettii Complex. PLoS Negl. Trop. Dis. 2014, 8, e3339. [Google Scholar] [CrossRef] [PubMed]
- Jariyapan, N.; Daroontum, T.; Jaiwong, K.; Chanmol, W.; Intakhan, N.; Sor-Suwan, S.; Siriyasatien, P.; Somboon, P.; Bates, M.D.; Bates, P.A. Leishmania (Mundinia) orientalis n. sp. (Trypanosomatidae), a parasite from Thailand responsible for localised cutaneous leishmaniasis. Parasites Vectors 2018, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Kwakye-Nuako, G.; Mosore, M.-T.; Duplessis, C.; Bates, M.D.; Puplampu, N.; Mensah-Attipoe, I.; Desewu, K.; Afegbe, G.; Asmah, R.H.; Jamjoom, M.B.; et al. First isolation of a new species of Leishmania responsible for human cutaneous leishmaniasis in Ghana and classification in the Leishmania enriettii complex. Int. J. Parasitol. 2015, 45, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Kwakye-Nuako, G.; Mosore, M.-T.; Boakye, D.; Bates, P.A. Description, biology, and medical significance of Leishmania (Mudinia) chancei n. sp. (kinetoplastea: Trypanosomatidae) from Ghana and Leishmania (Mudinia) procaviensis n. sp. (Kinetoplastea: Trypanpsomatidae) from Namibia. J. Parasitol. 2023, 109, 43–50. [Google Scholar] [CrossRef]
- Rose, K.; Curtis, J.; Baldwin, T.; Mathis, A.; Kumar, B.; Sakthianandeswaren, A.; Spurck, T.; Low Choy, J.; Handman, E. Cutaneous leishmaniasis in red kangaroos: Isolation and characterisation of the causative organisms. Int. J. Parasitol. 2004, 34, 655–664. [Google Scholar] [CrossRef]
- Dougall, A.M.; Alexander, B.; Holt, D.; Harris, T.; Sultan, A.H.; Bates, P.; Rose, K.; Walton, S. Evidence incriminating midges (Diptera: Ceratopogonidae) as potential vectors of Leishmania in Australia. Int. J. Parasitol. 2011, 41, 571–597. [Google Scholar] [CrossRef]
- Leelayoova, S.; Siripattanapipong, S.; Manomat, J.; Piyaraj, P.; Tan-Ariya, P.; Bualert, L.; Mungthin, M. Leishmaniasis in Thailand: A Review of Causative Agents and Situations. Am. J. Trop. Med. Hyg. 2017, 96, 534–542. [Google Scholar] [CrossRef]
- Srivarasat, S.; Brownell, N.; Siriyasatien, P.; Noppakun, N.; Asawanonda, P.; Rattanakorn, K.; Preativatanyou, K.; Kumtornrut, C. Case Report: Autochthonous Disseminated Cutaneous, Mucocutaneous, and Visceral Leishmaniasis Caused by Leishmania martiniquensis in a Patient with HIV/AIDS from Northern Thailand and Literature Review. Am. J. Trop. Med. Hyg. 2022, 107, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Anugulruengkitt, S.; Songtaweesin, W.N.; Thepnarong, N.; Tangthanapalakul, A.; Sitthisan, M.; Chatproedprai, S.; Wititsuwannakul, J.; Likitnukul, S.; Jariyapan, N.; Weedall, G.D.; et al. Case Report: Simple Nodular Cutaneous Leishmaniasis Caused by Autochthonous Leishmania (Mundinia) orientalis in an 18-Month-Old Girl: The First Pediatric Case in Thailand and Literature Review. Am. J. Trop. Med. Hyg. 2022, 108, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Cecílio, P.; Cordeiro-Da-Silva, A.; Oliveira, F. Sand flies: Basic information on the vectors of leishmaniasis and their interactions with Leishmania parasites. Commun. Biol. 2022, 5, 305. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, J.; Prasetyo, D.B.; Fiorenzano, J.M.; von Fricken, M.E.; Hertz, J.C. Phlebotomine sand flies (Diptera: Psychodidae) and sand fly-borne pathogens in the Greater Mekong Subregion: A systematic review. Parasites Vectors 2022, 15, 355. [Google Scholar] [CrossRef] [PubMed]
- Renaux Torres, M.C.; Pellot, C.; Somwang, P.; Khositharattanakool, P.; Vongphayloth, K.; Randrianambinintsoa, F.J.; Mathieu, B.; Siriyasatien, P.; Gay, F.; Depaquit, J. Phlebotomine sand flies (Diptera, Psychodidae) from Pha Tong cave, Northern Thailand with a description of two new species and taxonomical thoughts about Phlebotomus stantoni. PLoS Negl. Trop. Dis. 2023, 17, e0011565. [Google Scholar] [CrossRef] [PubMed]
- Buatong, J.; Dvorak, V.; Thepparat, A.; Thongkhao, K.; Koyadun, S.; Siriyasatien, P.; Pengsakul, T. Phlebotomine Sand Flies in Southern Thailand: Entomological Survey, Identification of Blood Meals and Molecular Detection of Trypanosoma spp. Insects 2022, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Preativatanyou, K.; Chinwirunsirisup, K.; Phumee, A.; Khositharattanakool, P.; Sunantaraporn, S.; Depaquit, J.; Siriyasatien, P. Species diversity of phlebotomine sand flies and sympatric occurrence of Leishmania (Mundinia) martiniquensis, Leishmania (Leishmania) donovani complex, and Trypanosoma spp. in the visceral leishmaniasis focus of southern Thailand. Acta Trop. 2023, 244, 106949. [Google Scholar] [CrossRef] [PubMed]
- Srisuton, P.; Phumee, A.; Sunantaraporn, S.; Boonserm, R.; Sor-Suwan, S.; Brownell, N.; Pengsakul, T.; Siriyasatien, P. Detection of Leishmania and Trypanosoma DNA in Field-Caught Sand Flies from Endemic and Non-Endemic Areas of Leishmaniasis in Southern Thailand. Insects 2019, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Chusri, S.; Thammapalo, S.; Silpapojakul, K.; Siriyasatien, P. Animal reservoirs and potential vectors of Leishmania siamensis in southern Thailand. Southeast Asian J. Trop. Med. Public Health 2014, 45, 13–19. [Google Scholar]
- Siripattanapipong, S.; Leelayoova, S.; Ninsaeng, U.; Mungthin, M. Detection of DNA of Leishmania siamensis in Sergentomyia (Neophlebotomus) iyengari (Diptera: Psychodidae) and Molecular Identification of Blood Meals of Sand Flies in an Affected Area, Southern Thailand. J. Med. Èntomol. 2018, 55, 1277–1283. [Google Scholar] [CrossRef]
- Sriwongpan, P.; Nedsuwan, S.; Manomat, J.; Charoensakulchai, S.; Lacharojana, K.; Sankwan, J.; Kobpungton, N.; Sriwongpun, T.; Leelayoova, S.; Mungthin, M.; et al. Prevalence and associated risk factors of Leishmania infection among immunocompetent hosts, a community-based study in Chiang Rai, Thailand. PLoS Negl. Trop. Dis. 2021, 15, e0009545. [Google Scholar] [CrossRef] [PubMed]
- Becvar, T.; Vojtkova, B.; Siriyasatien, P.; Votypka, J.; Modry, D.; Jahn, P.; Bates, P.; Carpenter, S.; Volf, P.; Sadlova, J. Exper-imental transmission of Leishmania (Mundinia) parasites by biting midges (Diptera: Ceratopogonidae). PLoS Pathog. 2021, 17, e1009654. [Google Scholar] [CrossRef] [PubMed]
- Seblova, V.; Sadlova, J.; Vojtkova, B.; Votypka, J.; Carpenter, S.; Bates, P.A.; Volf, P. The Biting Midge Culicoides sonorensis (Diptera: Ceratopogonidae) Is Capable of Developing Late Stage Infections of Leishmania enriettii. PLoS Negl. Trop. Dis. 2015, 9, e0004060. [Google Scholar] [CrossRef] [PubMed]
- Chanmol, W.; Jariyapan, N.; Somboon, P.; Bates, M.D.; Bates, P.A. Development of Leishmania orientalis in the sand fly Lutzomyia longipalpis (Diptera: Psychodidae) and the biting midge Culicoides soronensis (Diptera: Ceratopogonidae). Acta Trop. 2019, 199, 105157. [Google Scholar] [CrossRef] [PubMed]
- Wirth, W.W.; Hubert, A.A. The Culicoides of Southeast Asia (Diptera: Ceratopogonidae); Memoirs of the American Entomological Institute: Gainesville, FL, USA, 1989; Volume 44, pp. 1–508. [Google Scholar]
- Sick, F.; Beer, M.; Kampen, H.; Wernike, K. Culicoides Biting Midges-Underestimated Vectors for Arboviruses of Public Health and Veterinary Importance. Viruses 2019, 11, 376. [Google Scholar] [CrossRef] [PubMed]
- Rebêlo, J.M.M.; Rodrigues, B.; Bandeira, M.D.C.A.; Moraes, J.L.P.; Fonteles, R.S.; Pereira, S.R.F. Detection of Leishmania amazonensis and Leishmania braziliensis in Culicoides (Diptera, Ceratopogonidae) in an endemic area of cutaneous leishmaniasis in the Brazilian Amazonia. J. Vector Ecol. 2016, 41, 303–308. [Google Scholar] [CrossRef]
- Slama, D.; Haouas, N.; Remadi, L.; Mezhoud, H.; Babba, H.; Chaker, E. First detection of Leishmania infantum (Kinetoplastida: Trypanosomatidae) in Culicoides spp. (Diptera: Ceratopogonidae). Parasites Vectors 2014, 7, 51. [Google Scholar] [CrossRef]
- Ríos-Tostado, J.J.; Castillo-Ureta, H.; Torres-Montoya, E.H.; Torres-Avendaño, J.I.; Olimón-Andalón, V.; Romero-Higareda, C.E.; Silva-Hidalgo, G.; Zazueta-Moreno, J.M. Molecular Detection of Leishmania (L.) mexicana (Kinetoplastida: Trypanosto-matidae) DNA in Culicoides furens (Diptera: Ceratopogonidae) from an Area with Autochthonous Canine Leishmaniasis in Northwestern Mexico. Acta Parasitol. 2021, 66, 1055–1058. [Google Scholar] [CrossRef]
- Sunantaraporn, S.; Thepparat, A.; Phumee, A.; Sor-Suwan, S.; Boonserm, R.; Bellis, G.; Siriyasatien, P. Culicoides Latreille (Diptera: Ceratopogonidae) as potential vectors for Leishmania martiniquensis and Trypanosoma sp. in northern Thailand. PLoS Negl. Trop. Dis. 2021, 15, e0010014. [Google Scholar] [CrossRef]
- Dyce, A.L. The recognition of nulliparous and parous Culicoides (Diptera: Ceratopogonidae) without dissection. Aust. J. Entomol. 1969, 8, 11–15. [Google Scholar] [CrossRef]
- Pramual, P.; Jomkumsing, P.; Piraonapicha, K.; Jumpato, W. Integrative taxonomy uncovers a new Culicoides (Diptera: Ceratopogonidae) biting midge species from Thailand. Acta. Trop. 2021, 220, 105941. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; Ribeiro, G.C.; Cabral, A.D.; Sperança, M.A. A non-destructive enzymatic method to extract DNA from arthropod specimens: Implications for morphological and molecular studies. PLoS ONE 2018, 13, e0192200. [Google Scholar] [CrossRef] [PubMed]
- Manomat, J.; Leelayoova, S.; Bualert, L.; Tan-Ariya, P.; Siripattanapipong, S.; Mungthin, M.; Naaglor, T.; Piyaraj, P. Prevalence and risk factors associated with Leishmania infection in Trang Province, southern Thailand. PLoS Negl. Trop. Dis. 2017, 11, e0006095. [Google Scholar] [CrossRef] [PubMed]
- Coster, D.W.; Rademakers, R. NanoPack2: Population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef] [PubMed]
- Vierstraete, A.R.; Braeckman, B.P. Amplicon_sorter: A tool for reference-free amplicon sorting based on sequence similarity and for building consensus sequences. Ecol. Evol. 2022, 12, e8603. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Paradis, E. pagas: An R package for population genetics with an integrated−modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 April 2024).
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef]
- Phumee, A.; Tawatsin, A.; Thavara, U.; Pengsakul, T.; Thammapalo, S.; Depaquit, J.; Gay, F.; Siriyasatien, P. Detection of an Unknown Trypanosoma DNA in a Phlebotomus stantoni (Diptera: Psychodidae) Collected from Southern Thailand and Records of New Sand Flies With Reinstatement of Sergentomyia hivernus Raynal & Gaschen, 1935 (Diptera: Psychodidae). J. Med. Entomol. 2017, 54, 429–434. [Google Scholar]
- Jurado, V.; Laiz Trobajo, L.; Rodríguez Nava, V.; Boiron, P.; Hermosin, B.; Sanchez-Moral, S.; Saiz-Jimenez, C. Pathogenic and opportunistic microorganisms in caves. Int. J. Speleol. 2010, 39, 15–24. [Google Scholar] [CrossRef]
- Obame-Nkoghe, J.; Rahola, N.; Ayala, D.; Yangari, P.; Jiolle, D.; Allene, X.; Bourgarel, M.; Maganga, G.D.; Berthet, N.; Leroy, E.M.; et al. Exploring the diversity of blood-sucking Diptera in caves of Central Africa. Sci. Rep. 2017, 7, 250. [Google Scholar] [CrossRef] [PubMed]
- Collins, Á.B.; Mee, J.F.; Doherty, M.L.; Barrett, D.J.; England, M.E. Culicoides species composition and abundance on Irish cattle farms: Implications for arboviral disease transmission. Parasites Vectors 2018, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.Y.; Brostaux, Y.; Haubruge, E.; Francis, F. Larval development sites of the main Culicoides species (Diptera: Certopogonidae) in northern Europe and distribution of coprophilic species larvae in Belgian pastures. Vet. Parasitol. 2014, 205, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.Y.; Saegerman, C.; Losson, B.; Beckers, Y.; Haubruge, E.; Francis, F. Chemical composition of silage residues sus-taining the larval development of the Culicoides obsoletus/Culicoides scoticus species (Diptera: Ceratopogonidae). Vet. Parasitol. 2013, 191, 197–201. [Google Scholar] [CrossRef]
- Gomontean, B.; Vaisusuk, K.; Chatan, W.; Wongpakam, K.; Sankul, P.; Lachanthuek, L.; Mintara, R.; Thanee, I.; Pramual, P. Diversity, Abundance and Host Blood Meal Analysis of Culicoides Latreille (Diptera: Ceratopogonidae) from Cattle Pens in Different Land Use Types from Thailand. Insects 2023, 14, 574. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.D. Local dispersal of the biting-midge Culicoides brevitarsis Kieffer (Diptera, Ceratopogonidae) in southeastern Australia. Aust. J. Zool. 1987, 35, 559–573. [Google Scholar] [CrossRef]
- Ducheyne, E.; De Deken, R.; Bécu, S.; Codina, B.; Nomikou, K.; Mangana-Vougiaki, O.; Georgiev, G.; Purse, B.V.; Hendickx, G. Quantifying the wind dispersal of Culicoides species in Greece and Bulgaria. Geospat. Health 2007, 1, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Mignotte, A.; Garros, C.; Dellicour, S.; Jacquot, M.; Gilbert, M.; Gardès, L.; Balenghien, T.; Duhayon, M.; Rakotoarivony, I.; de Wavrechin, M.; et al. High dispersal capacity of Culicoides obsoletus (Diptera: Ceratopogonidae), vector of bluetongue and Schmallenberg viruses, revealed by landscape genetic analyses. Parasites Vectors 2021, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Songumpai, N.; Promrangsee, C.; Noopetch, P.; Siriyasatien, P.; Preativatanyou, K. First Evidence of Co-Circulation of Emerging Leishmania martiniquensis, Leishmania orientalis, and Crithidia sp. in Culicoides Biting Midges (Diptera: Ceratopogonidae), the Putative Vectors for Autochthonous Transmission in Southern Thailand. Trop. Med. Infect. Dis. 2022, 7, 379. [Google Scholar] [CrossRef]
- Jomkumsing, P.; Surapinit, A.; Saengpara, T.; Pramual, P. Genetic variation, DNA barcoding and blood meal identification of Culicoides Latreille biting midges (Diptera: Ceratopogonidae) in Thailand. Acta Trop. 2021, 217, 105866. [Google Scholar] [CrossRef]
- Kamyingkird, K.; Choocherd, S.; Chimnoi, W.; Klinkaew, N.; Kengradomkij, C.; Phoosangwalthong, P.; Thammasonthijarern, N.; Pattanatanang, K.; Inpankaew, T.; Phasuk, J.; et al. Molecular Identification of Culicoides Species and Host Preference Blood Meal in the African Horse Sickness Outbreak-Affected Area in Hua Hin District, Prachuap Khiri Khan Province, Thailand. Insects 2023, 14, 369. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Mondal, B.; Pal, A.; Harsha, R.; Mazumdar, A. Blood meal analysis of Culicoides species associated with livestock in West Bengal, India. Med. Vet. Entomol. 2022, 36, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Sunantaraporn, S.; Hortiwakul, T.; Kraivichian, K.; Siriyasatien, P.; Brownell, N. Molecular Identification of Host Blood Meals and Detection of Blood Parasites in Culicoides Latreille (Diptera: Ceratopogonidae) Collected from Phatthalung Province, Southern Thailand. Insects 2022, 13, 912. [Google Scholar] [CrossRef] [PubMed]
- Hitakarun, A.; Tan-ariya, P.; Siripattanapipong, S.; Mungthin, M.; Piyaraj, P.; Naaglor, T.; Siriyasatien, P.; Tiwananthagorn, S.; Leelayoova, S. Comparison of PCR methods for detection of Leishmania siamensis infection. Parasites Vectors 2014, 7, 458. [Google Scholar] [CrossRef] [PubMed]
- Ruang-Areerate, T.; Ruang-Areerate, P.; Manomat, J.; Naaglor, T.; Piyaraj, P.; Mungthin, M.; Leelayoova, S.; Siripattanapipong, S. Genetic variation and geographic distribution of Leishmania orientalis and Leishmania martiniquensis among Leishmania/HIV co-infection in Thailand. Sci. Rep. 2023, 13, 23094. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, Z.; Azarang, E.; Shahabi, S.; Omidian, M.; Pourabbas, B.; Sarkari, B. Leishmania ITS1 Is Genetically Divergent in Asymptomatic and Symptomatic Visceral Leishmaniasis: Results of a Study in Southern Iran. J. Trop. Med. 2020, 2020, 5351098. [Google Scholar] [CrossRef]
- Chen, Y.F.; Liao, L.F.; Wu, N.; Gao, J.M.; Zhang, P.; Wen, Y.Z.; Hide, G.; Lai, D.H.; Lun, Z.R. Species identification and phylogenetic analysis of Leishmania isolated from patients, vectors and hares in the Xinjiang Autonomous Region, The People’s Republic of China. PLoS Negl. Trop. Dis. 2021, 15, e0010055. [Google Scholar] [CrossRef] [PubMed]
- Spotin, A.; Rouhani, S.; Haghighi, A.; Parvizi, P. Low genetic heterogeneity of Leishmania major in different geographical regions of Iran. PLoS ONE 2023, 18, e0285520. [Google Scholar] [CrossRef]
- Charyyeva, A.; Çetinkaya, Ü.; Özkan, B.; Şahin, S.; Yaprak, N.; Şahin, I.; Yurchenko, V.; Kostygov, A.Y. Genetic diversity of Leishmania tropica: Unexpectedly complex distribution pattern. Acta Trop. 2021, 218, 105888. [Google Scholar] [CrossRef]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. Genetics 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Ferreri, M.; Qu, W.; Han, B. Phylogenetic networks: A tool to display character conflict and demographic history. Afr. J. Biotechnol. 2011, 10, 12799–12803. [Google Scholar]
- Ramos-Onsins, S.E.; Rozas, J. Statistical Properties of New Neutrality Tests Against Population Growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Cañeda-Guzmán, I.C.; Oca-Aguilar, A.C.M.; Miranda-Caballero, C.I.; Grostieta, E.; Correa-Morales, F.; Romero-Pérez, R.; Romero-Contreras, F.E.; Rodríguez-Atanacio, J.A.; Ruiz-Tovar, K.; Huerta, H.; et al. Entomological Survey and Leishmania (Leishmania) mexicana Prevalence in Sand Fly Species during an Outbreak of Cutaneous Leishmaniasis in Quintana Roo State, Mexico. Trop. Med. Infect. Dis. 2023, 8, 465. [Google Scholar] [CrossRef]
- Chiewchanvit, S.; Tovanabutra, N.; Jariyapan, N.; Bates, M.D.; Mahanupab, P.; Chuamanochan, M.; Tantiworawit, A.; Bates, P.A. Chronic generalized fibrotic skin lesions from disseminated leishmaniasis caused by Leishmania martiniquensis in two patients from northern Thailand infected with HIV. Br. J. Dermatol. 2015, 173, 663–670. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Sites | Genus (Subgenus/Species Group) | Species | No. Males | No. Non-Engorged Females | No. Females with Parasites Detected | Midge Specimen Vouchers with Parasite Identification | ||
|---|---|---|---|---|---|---|---|---|
| Parous | Gravid | L. martiniquensis | L. orientalis | |||||
| Patient Residence Mae Kon, Muang District Chiang Rai Province (19°51′05.8″ N, 99°39′37.1″ E) | C. (Hoffmania) | insignipennis | 2 | 15 | ||||
| C. (Hoffmania) | malayae | 6 | ||||||
| C. (Hoffmania) | liui | 5 | ||||||
| C. (Hoffmania) | innoxius | 5 | ||||||
| C. (Hoffmania) | sumatrae | 2 | ||||||
| C. (Trithecoides) | spp. | 5 | 17 | 1 | 8 | PR3, PR5, PR6, PR8, PR10, PR11, PR15, PR24 | PR3, PR5, PR11, PR24 | |
| C. (Avaritia) | orientalis | 12 | 1 | PR2 | PR2 | |||
| C. (Avaritia) | jacobsoni | 4 | 2 | PR21, PR23 | PR23 | |||
| C. (Remmia) | oxystoma | 2 | ||||||
| C. (Shermani group) | thurmanae | 1 | ||||||
| C. (Meijerehelea) | mahasarakhamense | 1 | 1 | PR22 | PR22 | |||
| Total | 7 | 67 | 4 | 12/71 (16.9%) | 12/71 (16.9%) | 7/71 (9.9%) | ||
| Grocery Shop Mae Kon, Muang District Chiang Rai Province (19°50′56.3″ N, 99°39′50.1″ E) | C. (Remmia) | oxystoma | 3 | 2 | GS10, GS12 | GS10, GS12 | ||
| C. (Avaritia) | jacobsoni | 2 | ||||||
| C. (Meijerehelea) | mahasarakhamense | 1 | 1 | GS1 | ||||
| Total | 6 | 3/6 (50%) | 3/6 (50%) | 2/6 (33.3%) | ||||
| Tham Phra Cave Mae Yao, Muang District Chiang Rai Province (19°55′03.5″ N, 99°47′19.5″ E) | C. (Meijerehelea) | guttifer | 7 | 34 | 7 | 11 | TP34, TP38, TP39, TP48, TP53, TP56, TP58, TP61, TP62, TP63, TP65 | TP34, TP38, TP48, TP53, TP63, TP65 |
| C. (Meijerehelea) | mahasarakhamense | 2 | 16 | 12 | 13 | TP33, TP43, TP45, TP47, TP49, TP50, TP51, TP54, TP55, TP57, TP59, TP60, TP64 | TP33, TP45, TP51, TP54, TP57, TP59, TP64 | |
| Total | 9 | 50 | 19 | 24/69 (34.8%) | 24/69 (34.8%) | 13/69 (18.8%) | ||
| Grand total | 16 | 146 | 39/146 (26.7%) | 39/146 (26.7%) | 22/146 (15.1%) | |||
| Parasite Population | Total Sample Size | No. Haplotypes (H) | No. Polymorphic Sites (S) | Average no. of Nucleotide Differences (k) | Haplotype Diversity (Hd) ± SD | Nucleotide Diversity (π) ± SD | Tajima’s D | Fu and Li’s D |
|---|---|---|---|---|---|---|---|---|
| L. martiniquensis from Culicoides (45) and patients (4) | 49 | 13 | 18 | 3.2179 | 0.4634 ± 0.0903 | 0.0041 ± 0.0031 | −2.3065 * | −3.0583 * |
| L. orientalis from Culicoides (22) and one patient (1) | 23 | 17 | 24 | 2.1618 | 0.9489 ± 0.0326 | 0.0059 ± 0.0041 | −2.8127 ** | −2.6740 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ampol, R.; Somwang, P.; Khositharattanakool, P.; Promrangsee, C.; Pataradool, T.; Tepboonreung, P.; Siriyasatien, P.; Preativatanyou, K. Nanopore-Based Surveillance of Leishmania Parasites in Culicoides Latrielle (Diptera: Ceratopogonidae) Caught from the Affected Community and Tham Phra Cave in Chiang Rai Province, the Endemic Area of Leishmaniasis in Northern Thailand. Insects 2024, 15, 327. https://doi.org/10.3390/insects15050327
Ampol R, Somwang P, Khositharattanakool P, Promrangsee C, Pataradool T, Tepboonreung P, Siriyasatien P, Preativatanyou K. Nanopore-Based Surveillance of Leishmania Parasites in Culicoides Latrielle (Diptera: Ceratopogonidae) Caught from the Affected Community and Tham Phra Cave in Chiang Rai Province, the Endemic Area of Leishmaniasis in Northern Thailand. Insects. 2024; 15(5):327. https://doi.org/10.3390/insects15050327
Chicago/Turabian StyleAmpol, Rinnara, Puckavadee Somwang, Pathamet Khositharattanakool, Chulaluk Promrangsee, Thanapat Pataradool, Piyapat Tepboonreung, Padet Siriyasatien, and Kanok Preativatanyou. 2024. "Nanopore-Based Surveillance of Leishmania Parasites in Culicoides Latrielle (Diptera: Ceratopogonidae) Caught from the Affected Community and Tham Phra Cave in Chiang Rai Province, the Endemic Area of Leishmaniasis in Northern Thailand" Insects 15, no. 5: 327. https://doi.org/10.3390/insects15050327