Using Entropy Leads to a Better Understanding of Biological Systems


Department of Oncology, University of Alberta, 11560 University Ave. Edmonton, AB T6G 1Z2, Canada
*
Author to whom correspondence should be addressed.
Entropy 2010, 12(12), 2450-2469; https://doi.org/10.3390/e12122450
Submission received: 4 November 2010
/
Revised: 7 December 2010
/
Accepted: 15 December 2010
/
Published: 17 December 2010
(This article belongs to the Special Issue Emergence in Chemical Systems)
Abstract
:In studying biological systems, conventional approaches based on the laws of physics almost always require introducing appropriate approximations. We argue that a comprehensive approach that integrates the laws of physics and principles of inference provides a better conceptual framework than these approaches to reveal emergence in such systems. The crux of this comprehensive approach hinges on entropy. Entropy is not merely a physical quantity. It is also a reasoning tool to process information with the least bias. By reviewing three distinctive examples from protein folding dynamics to drug design, we demonstrate the developments and applications of this comprehensive approach in the area of biological systems.
PACS Codes:
87.10.Ca; 87.15.hm; 87.19.xj; 87.85.fk1. Introduction
The methods of statistical mechanics have been favored by physicists investigating biological problems, which involve various emergent properties such as protein folding dynamics and protein-protein interactions in drug discovery. However, due to the presence of many-body interactions in such systems, applications of these methods made without appropriate approximations result in intractable problems [1]. For example, in studying fluid structures in physics, Tseng and Caticha [1] discuss this issue and give an example of seeking out optimal hard sphere approximations to properly describe many-body interactions. Similarly, in studying protein structure in biology, the lattice model is introduced to approximate complicated atomic interactions within proteins [2,3].
We argue that an alternative and perhaps more comprehensive solution to introducing approximations is that suggested by Wheeler [4], who concluded that all things physical are information-theoretic in origin and we live in a participatory universe. This suggests a comprehensive route, in which one integrates physics and information to develop reasoning and to create theories instead of devising approximations that might be appropriate for biological systems. Because the studies of information theory focus on applying principles of inference to consistently, objectively, universally, and honestly process information, the key to integrating physics and information is the same as that required for integrating the laws of physics and principles of inference. Furthermore, because the concept of entropy is not only found to represent irreversibility in thermodynamics but has also been shown to be the foundation of information processing [5,6,7,8,9] and is a means for deriving the dynamics of statistical systems purely from the principles of inference [10,11,12,13], we conclude that the key to this type of integration is rooted in entropy.
Here, we review three distinctive examples in biology and drug discovery to demonstrate the developments, applications and advantages of the approach based on this comprehensive route and to reveal emergence in these complex systems. In the first example, we will show that the incorporation of the laws of physics and principles of inference provides a more comprehensive method to investigate and reveal protein folding dynamics compared to conventional approaches [14]. Second, a maximum entropy method is developed to predict tubulin isotype expression levels in a cell when the cell is exposed to various cytotoxic derivatives of the anti-cancer drug, colchicine [15]. The last example discussed here aims to provide a theoretical method based on the maximum entropy approach to design short nucleic acid sequences, aptamers that specifically bind to bio-molecular targets of interest [16]. These three examples will provide strong evidence that entropy plays a crucial and fundamental role in conceptual development rather than being merely involved in either a measurement of randomness or a tool for processing information.
2. A Comprehensive Perspective
2.1. The Core Aspect: Rules of Probability Theory, the Method of Maximum Entropy and Information Geometry
To formulate a comprehensive approach that integrates physics and information for the study of biological systems, we consider a procedure that is different from which that is the conventionally used in the past to set up physical theories [6]. Normally one starts by establishing a mathematical formalism, and then one tries to append an interpretation to it. The alternative procedure works in the opposite direction, one starts by determining what is the object of investigation and the goals that are being set, and only afterward one designs an appropriate mathematical formalism.
An appropriate formalism for this alternative procedure involves the principles of inference, in which probability is considered to be the degree of belief or plausibility rather than merely a frequency. The principles of inference including consistency, objectivity, universality, and honesty, are sufficiently constraining that they lead to a unique set of rules for processing information: rules of probability theory [17,18,19] and the method of maximum entropy [5,6,7,8,9,20]. Furthermore, it was found that information geometry is a convenient tool to manipulate information [21]. A brief illustration of three specific methods is given below.
Rules of probability theory. Cox showed that the Bayesian interpretation of probability and entropy can also be manipulated by the rules of subtraction, multiplication and addition of standard probability theory [17,18]. Namely, one can quantitatively manipulate propositions regarding systems of interest [17,18,19].
Method of maximum entropy. Following the alternative route to solve statistical problems, entropy is just a tool for induction [6]. First, Jaynes showed that the method of maximum entropy (denoted by MaxEnt) is a reasoning tool to assign probabilities on the basis of limited information with the least bias [5,22]. The preferred probability distribution of the system in a specific state is the one that maximizes the entropy of the system subject to the information available. Second, Shore and Johnson, Caticha and Giffin followed the same reasoning and proposed that the method of maximum entropy (denoted by ME) can also be utilized as a tool for updating probability distributions based on limited information [5,6,7,8,9,20]. They showed that the key to updating probability distributions is through relative entropy rather than the entropy in MaxEnt. Note that relative entropy is defined as the negative Kullback-Leibler distance of two probability distributions and quantifies their difference. Therefore, to update from a probability distribution, which we call a prior distribution, based on available new information to some probability distributions, defined as posterior distributions, with the least bias, one needs to maximize the relative entropy of all possible distributions and the prior distribution subject to this new information. Subsequent maximization will then lead to the preferred posterior distribution.
Information geometry. Based on the method of differential geometry, information geometry introduces the concept of manifolds for characterizing and manipulating information [21]. An information manifold is constructed based on independent parameters that characterize the system of interest. The probability distributions of the system in specific states can then be treated as points in the manifold. Fisher and Rao showed a uniquely natural way to quantify changes between the two points, which is now recognized as the Fisher-Rao metric [23,24,25]. Therefore, when a probability is assigned to each point, it automatically provides the space of states with a metric structure. Consequently, all information regarding the systems of interest is embedded in the corresponding information manifolds.
2.2. Comments
In this alternative approach to study biological systems, one first focuses on finding out what the “right” questions and goals are and then designs appropriate mathematical formalisms. It is found that such mathematical formalisms are based on the rules of probability theory, the method of maximum entropy and information geometry. This way of formulating theories provides an advantage that conventional approaches lack in investigating biological systems. Namely, we are utilizing information relevant to systems and problems of interest that is acquired from the applications of laws of physics to make best inferences following the principles of inference in order to answer our questions rather than attempting to solve some mathematical equations and appending interpretations to it. Suppose there exists an exact formalism to interpret phenomena pertinent to biological systems. Since the principles of inference represent rigorous and objective ways of processing information, the formalism resulting from this alternative approach is likely to be closest to such an exact formalism if sufficient information and appropriate laws of physics are utilized. In the following section, we demonstrate the developments and applications stemming from this approach using three examples to solve problems that are difficult to answer using conventional approaches.
3. From Principles of Inference to Protein Folding Dynamics
3.1. Introduction
Protein folding dynamics is one of the major issues investigated in the study of protein functions. Because the protein folding process involves complicated many-body interactions, molecular dynamics (MD) simulations are a common theoretical/computational approach considered. However, one issue hinders the practical usage of MD simulations in studying protein folding processes. Proteins may be trapped in one of many local energy minima on the energy surface during simulations. To resolve this issue, the replica exchange method (REM) has been proposed [26]. Yet, the introduction of the Monte Carlo aspect in REM seems to inevitably lead to the loss of the dynamical information about the folding process. In Tseng et al. [14], we proposed to tackle the folding dynamics problem by asking the following question: “Can we directly reveal folding dynamics from pure REM-MD simulation results? And if so, how?”
3.2. Theory
In trying to answer the question: “Can laws of physics be derived from principles of inference?”, the work of Caticha and collaborators [10,11,12] unveils a solution to this problem. They argue that information geometry is a convenient tool to utilize in order to proceed. Based on information geometry, a natural way to distinguish two macrostates Aα and Aα+dAα is to treat each as a point in the space of states, the information manifold with coordinates Aα. One can then show that the difference between the two states is given by the distance dl between p(x| Aα) and p(x| Aα+dAα), namely:
where a general expression of gαβ is given by:
which is the Fisher-Rao metric [23,24,25]. This is the only Riemannian metric that adequately reflects the underlying statistical nature of the manifold of distributions p(x|A). This result indicates that when the probability p(x|Aα) is assigned to each point Aα, it automatically provides the space of states with a metric structure. Note that the coordinates of the manifold need not be the expectation values. Because we chose the expectation values Aα as the coordinates, one can show that an alternative expression for the Fisher-Rao metric is:
The evolution of probability distributions is then simply represented by a point object that “moves” along a trajectory within the manifold. Based on this treatment, Caticha showed that the dynamics of a physical system can be derived directly from principles of inference. He, therefore, termed this approach the entropic dynamics. In entropic dynamics, dynamical laws follow from recognizing the appearance of changes from one point to a neighboring point in the manifold. According to the ME principle, the preferred neighboring point is the one corresponding to the state of maximum entropy.
Following the studies of Caticha [10,11,12], we have argued that protein folding dynamics can also be derived from entropic dynamics given REM-MD simulation results of a target protein, which forms a statistical system of possible protein structures, and have proposed entropic folding dynamics [14]. The first step in entropic folding dynamics is to construct the information manifold of the statistical system that codifies structural and dynamical information of a protein. Specifically, some quantities such as energy or the root-mean-square deviation (RMSD) of two structures from the same REM-MD results that characterize protein structures and the properties of slow folding process can be considered to be fundamental quantities used to construct coordinates of the information manifold. Next, suppose all possible folding trajectories of that protein can be described by those quantities. We can then treat a transition state along the folding trajectories as an average structure of all possible structures in the phase space with specific probabilities, P(i), where “I” denotes the ith microstate of this protein’s statistical system which represents a protein structure at a specific time and temperature in the simulations.
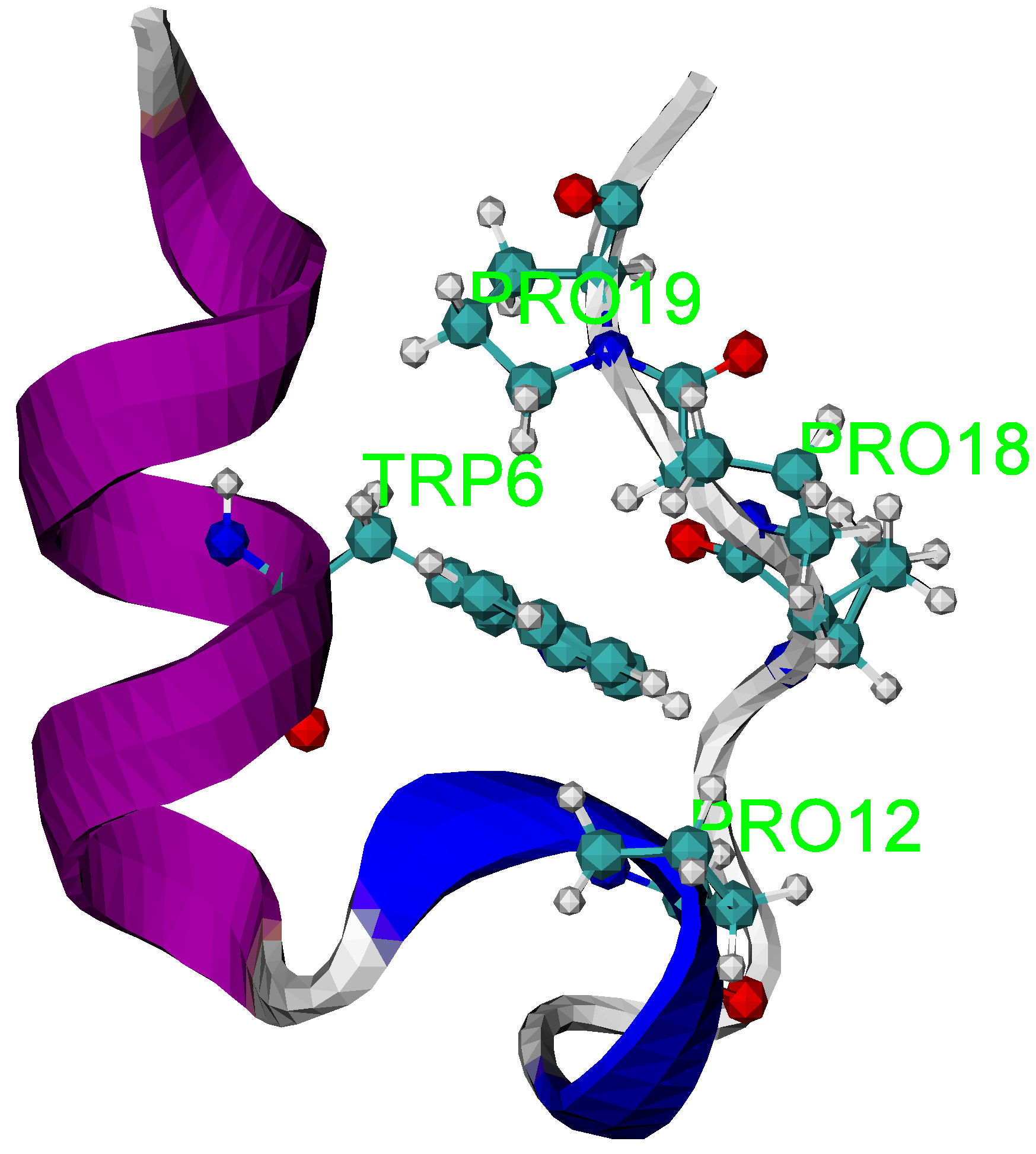
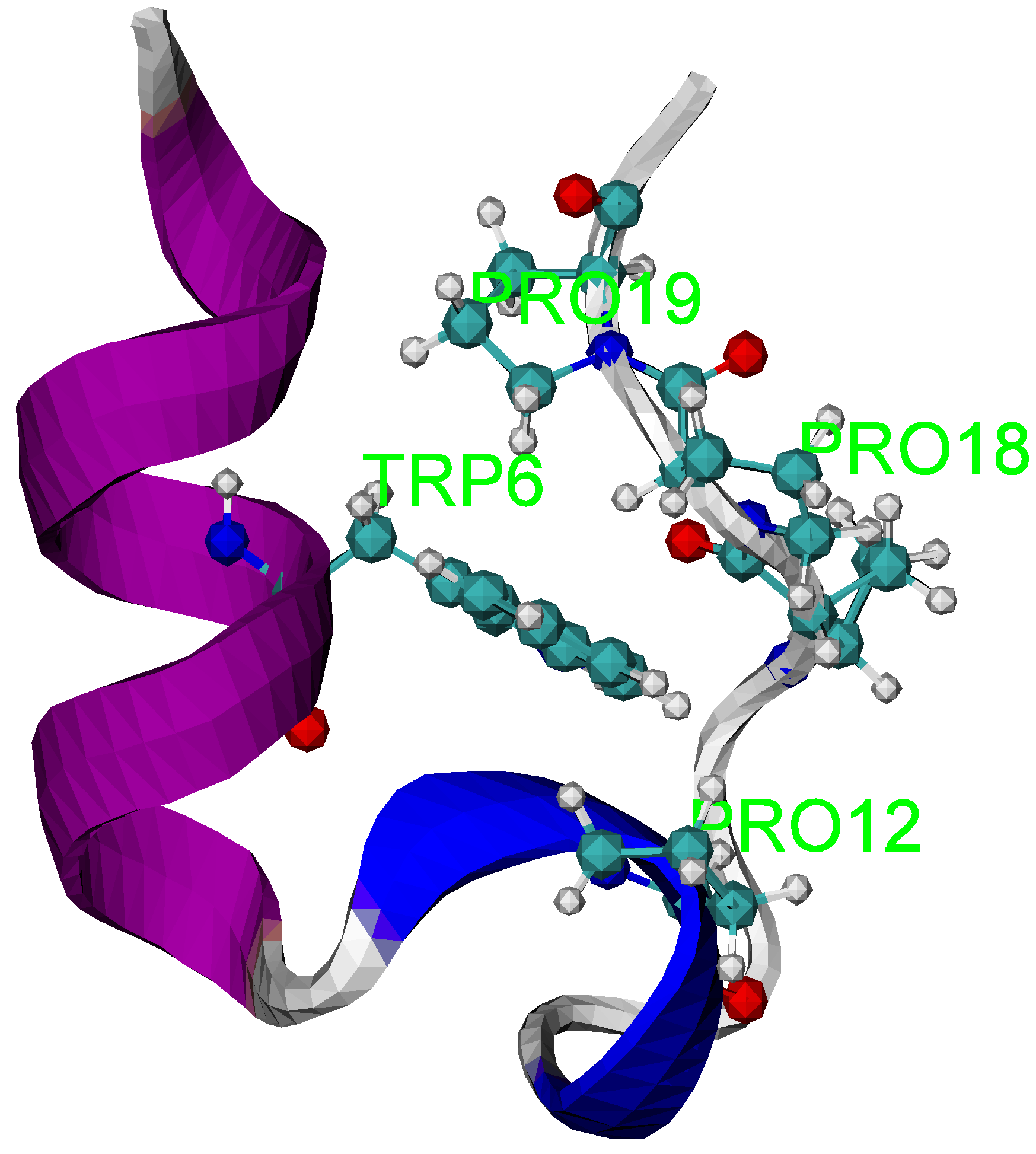
In Tseng et al. [14], a well-studied Trp-cage peptide, the native structure is shown in Figure 1 generated by the software package, visual molecular dynamics (VMD) [27], which illustrates the applicability of the entropic folding dynamics.
Figure 1.
The NMR structure of the Trp cage (PDB id: 1L2Y). The structure shows Trp6 is caged inside the hydrophobic pocket formed by three proline residues.
Figure 1.
The NMR structure of the Trp cage (PDB id: 1L2Y). The structure shows Trp6 is caged inside the hydrophobic pocket formed by three proline residues.
Details of the REM-MD simulations to generate Trp-cage statistical system are given in [14]. To construct the information manifold, we consider two quantities. The first quantity is the reachability distance that quantifies structural differences through ranking proteins in the order of similarity, which is defined by RMSD of two structures, without introducing any reference structures using a density-based clustering analysis method, Ordering Points to Identify the Clustering Structure (OPTICS) [28]. The second quantity is the smoothed potential energy that characterizes slow folding processes, which is obtained by removing an energy component corresponding to fast atomic motions. Therefore, we define two macrostates:
where N is the total number of protein structures, B¹ denotes the expectation reachability distance and B² is the expectation smoothed potential energy as the two coordinates for the information manifold. Note that ideally, all possible structures in the folding process are expected to be equally generated from the REM-MD simulations. When there are no constraints provided, the most honest choice is to assign an equal probability to each structure as a prior. Furthermore, even though one of the constraints, smoothed potential energy, refers to a quasi-static equilibrium state, Jaynes proved that the method of maximum entropy is still applicable [29]. Therefore, given a uniform prior probability and the constraint equation, Equation (4), the ME probability distribution function is given by:
where the partition function is . Note that in tensor notation, , is used. The Lagrangian multipliers γα are determined numerically. Furthermore, the entropy of the Trp-cage statistical system in state Bα is given by:
Finally, according to information geometry, when a probability distribution is defined, a metric space for the information manifold is created naturally. Therefore, the action of choosing the expectation reachability-distance and smoothed potential energy as the coordinate system defines the metric tensor, shown in Equation (3).
3.3. Folding Trajectories of Trp-Cage
After the metric space is determined, the evolution of the Trp-cage from a given initial macrostate to the final macrostate in the information manifold is determined by the ME principle. The Trp-cage statistical system will evolve from the jth macrostate Bα(j) to a neighboring state Bα(j + 1) via:
The change, dBα(j) calculated from Equation (1), is:
where γα(j) = gαβ(j)γβ(j) is the Lagrangian multiplier γα at jth step and gαβ(j)gαβ(j) = 1. However, Equation (7) is not yet a physical law without introducing dynamical information to constrain dl, the minimum distance between two macrostates. To constrain dl, as is recognized in many studies, proteins tend to fold in a direction that globally decreases the reachability distance and potential energy. Therefore, we propose to constrain the “direction” of changes with regard to the potential energy and reachability distance by setting a negative absolute speed dl/dt when the rate of the jth macrostate change dBα(j)/dl is positive [14].
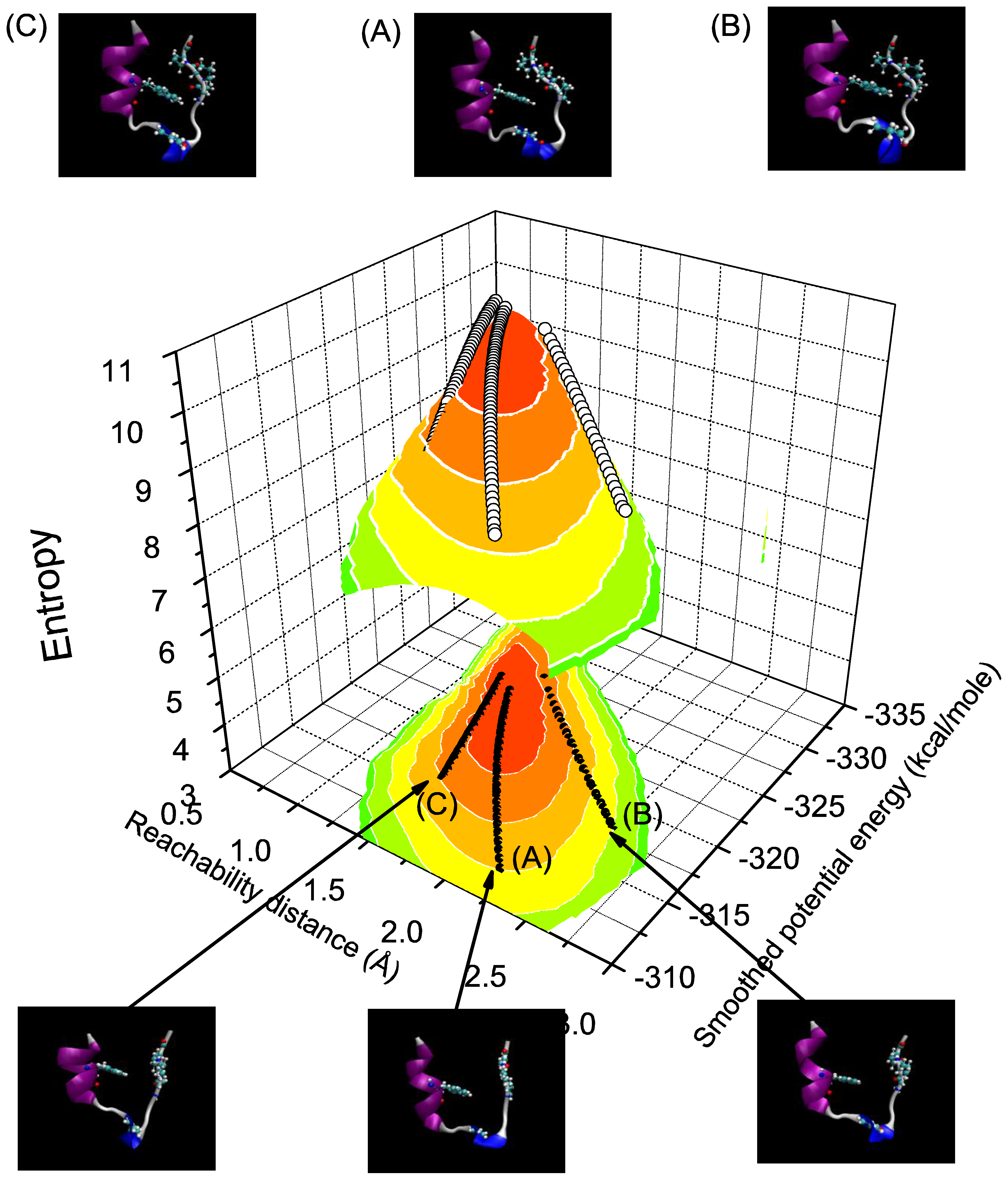
We investigate the properties of the folding trajectories when the initial states are given differently with dl/dt = 0.01. The three initial states are: (A) B¹ = 2.2Å; B² = −312 kcal/mole, (B), B¹ = 2.5 Å; B² = −318, and (C) B¹ = 1.5 Å; B² = −316 kcal/mole. The initial state (B) has the lowest smoothed potential energy, and yet has the largest reachability distance. In contrast, the initial state (C) has the shortest reachability distance. Both initial states are in the vicinity of the upper and lower boundaries, respectively, of the sampling space generated from the REM-MD simulation results. Figure 2 shows how the Trp-cage evolves toward the maximum entropy state on the three-dimensional entropy surface through three trajectories. It also shows the steepness of this entropy surface. Furthermore, we project the same trajectories onto the two-dimensional entropy contour map in the bottom to show the differences between the trajectories in terms of the smoothed potential energy and reachability distance. Six cartoon representations of the averaged Trp-cage structure at initial and final steps are presented in the same figure. The entropy of the system is calculated from Equation (6) for each state Bα and its magnitudes are denoted by the color scale. Note that the entropy contour map is plotted to roughly cover the region of the sampling space. It takes 57, 29 and 48 steps for state (A), (B) and (C) to reach the maximum entropy state, respectively.
The figure shows two features of folding trajectories. The first feature, revealed by either trajectory (B) or (C), is that the system likely evolves through a straight route. There is a linear relation between the smoothed potential energy and reachability distance. The second feature, revealed by trajectory (A), shows a curved type route. This type of route first shows that the system evolves with the reachability distance having a decreasing rate which is faster than the smoothed potential energy rate. After around B¹ = 1.75 Å; B² = −316 kcal/mole, the system evolves with the decreasing rate for the reachability distance being slower than the decreasing rate for the smoothed potential energy. In general, when the initial state of the system is in the vicinity of the sampling space, the system tends to evolve through a straight route. Otherwise, a curved type route will be expected.
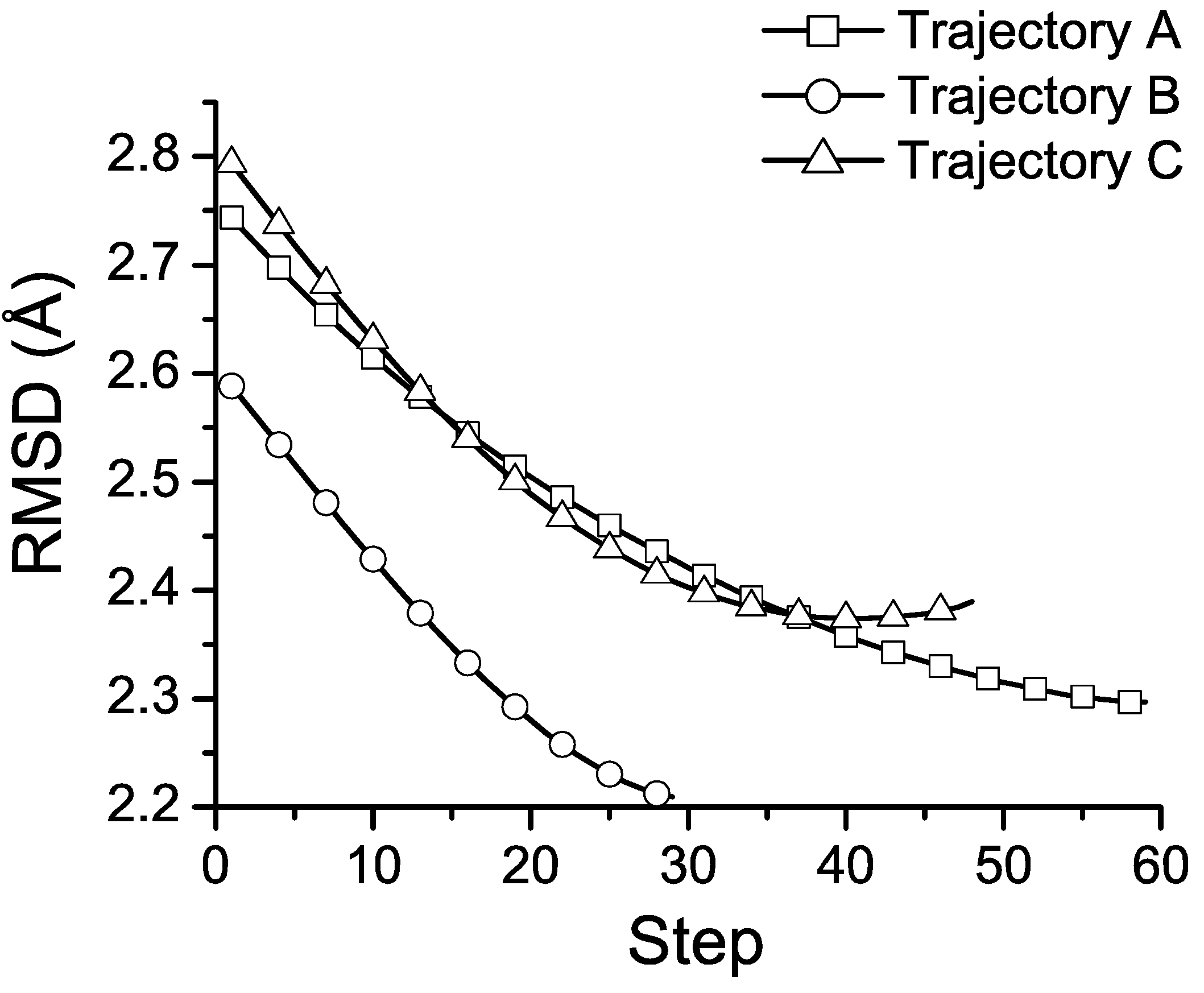
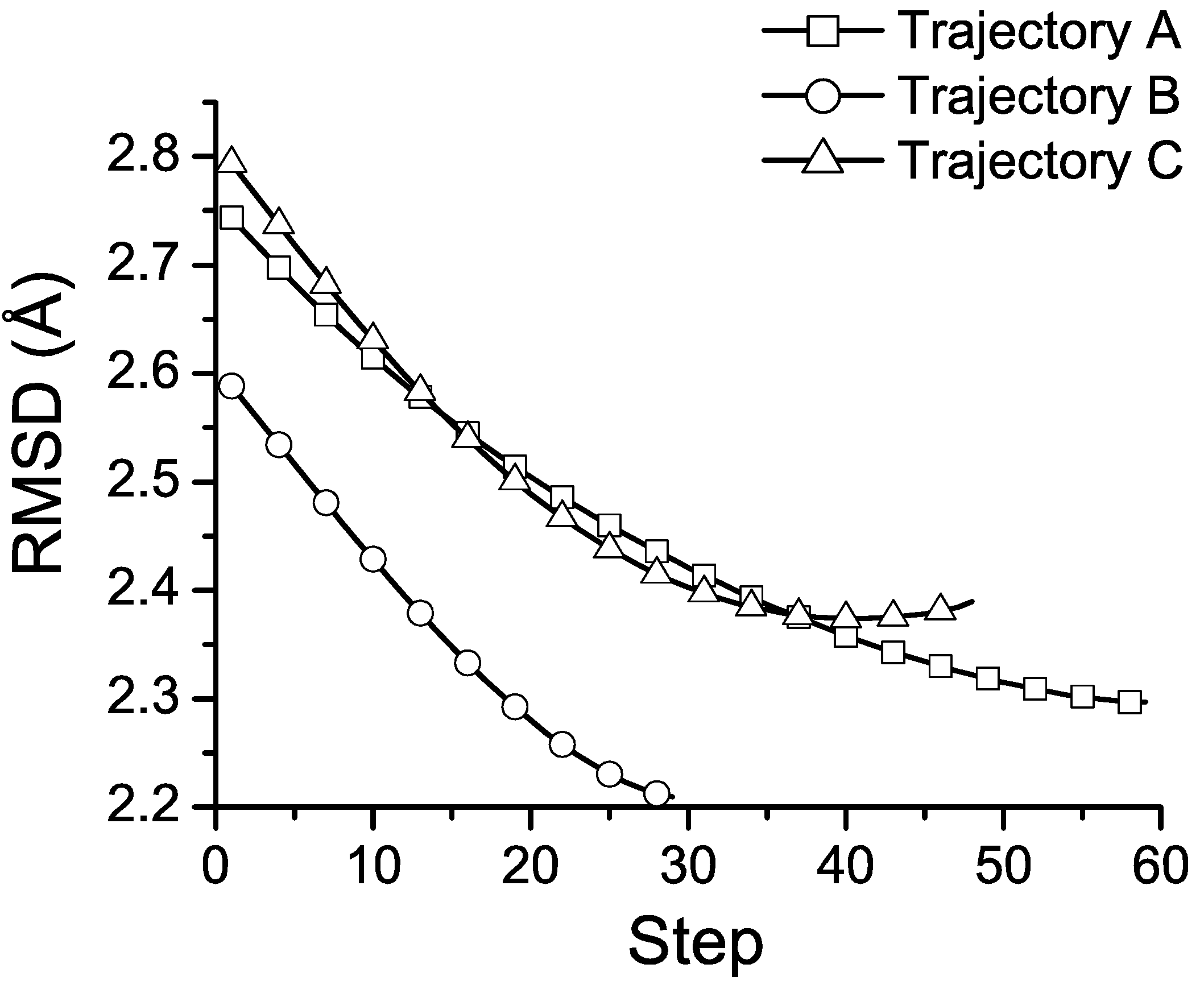
To quantitatively analyze the structural changes along the trajectories, we calculate the RMSD of the Cα-atoms of the average structures at each step of the three trajectories and the NMR structure of the Trp-cage. The results are shown in Figure 3. The RMSD of the final structures and the NMR structure are 2.17, 2.17, and 2.26 Å, respectively. For both trajectories A and B, the RMSD values gradually decrease to around 2.13 Å ~after 45 and 20 steps and climb back a little to 2.17 Å, respectively. This suggests that when the system evolves to the lower left region in Figure 2, the corresponding average structure is likely to be equilibrated and approaches the native structure.
Figure 2.
Entropic folding trajectories of the Trp-cage given three various initial structures. The three trajectories are plotted in the three-dimensional entropy space. In the bottom part of the figure, the same trajectories are projected on the two-dimensional entropy contour map. The averaged initial (bottom) and final (top) structures generated by VMD [27] are also presented for comparison.
Figure 2.
Entropic folding trajectories of the Trp-cage given three various initial structures. The three trajectories are plotted in the three-dimensional entropy space. In the bottom part of the figure, the same trajectories are projected on the two-dimensional entropy contour map. The averaged initial (bottom) and final (top) structures generated by VMD [27] are also presented for comparison.
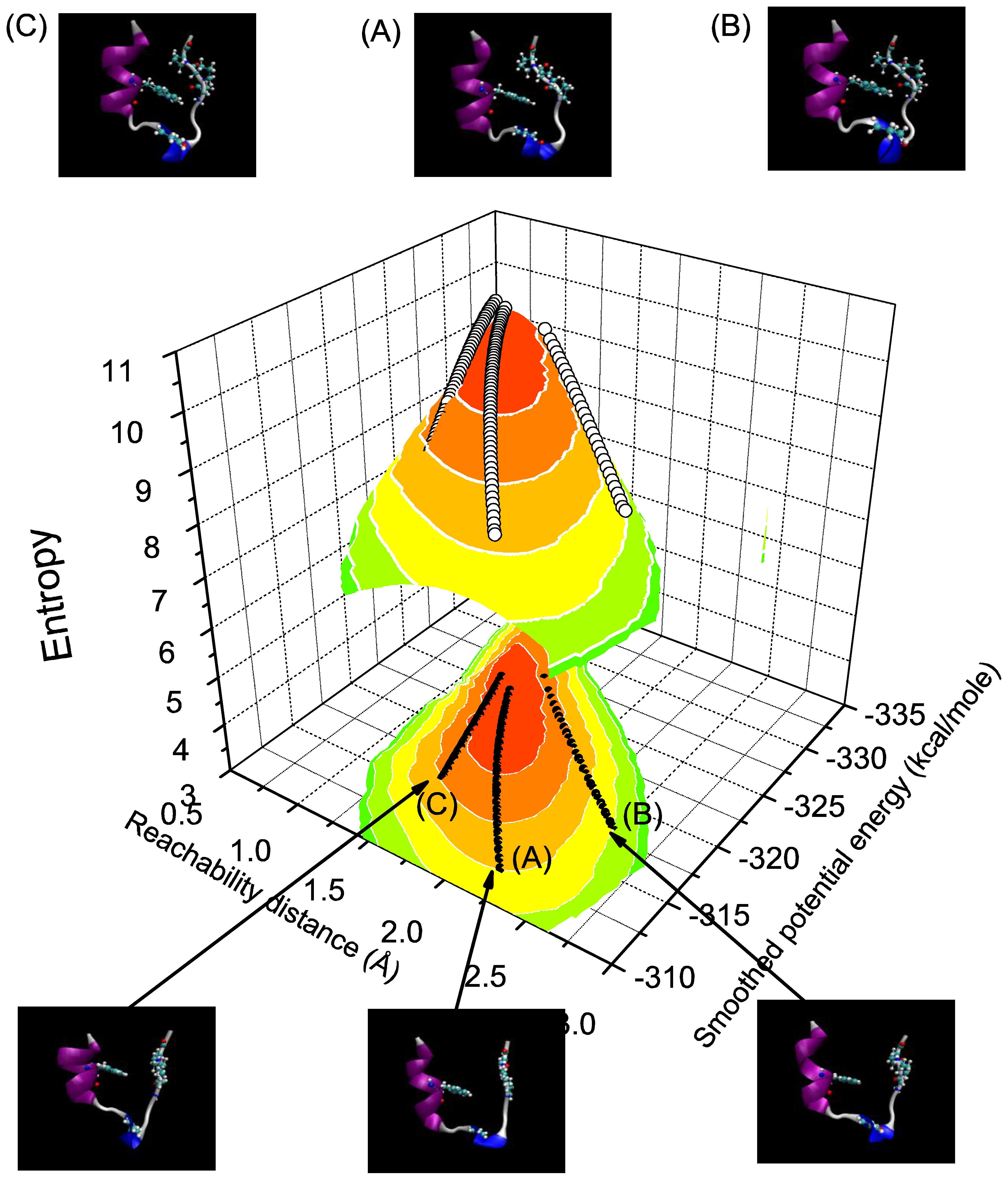
Figure 3.
The RMSD values of averaged structures along the three trajectories and the NMR structure of the Trp-cage. The lines with hollow squares, circles and triangles are from trajectories (A), (B) and (C), respectively. Note that we only label the data points by symbols at every 4 steps.
Figure 3.
The RMSD values of averaged structures along the three trajectories and the NMR structure of the Trp-cage. The lines with hollow squares, circles and triangles are from trajectories (A), (B) and (C), respectively. Note that we only label the data points by symbols at every 4 steps.
3.4. Discussion
The entropic folding process qualitatively agrees with the results of other studies such as Juraszek and Bolhuis [30], in which the transition pathway sampling technique [31,32,33] is integrated in MD simulations, to solve the sampling issue in order to study the folding dynamics. Our studies also present one advantage and give an insight with regard to the folding dynamics. The advantage is that there is no need to modify the current REM-MD simulation protocol, such as by integrating it with some sampling techniques. Instead, one can directly apply the proposed approach to the systems that have been investigated using either REM-MD or Monte Carlo simulations as long as the “right” coordinate systems are defined to construct the information manifold. Regarding the new insight, our results show that the folding dynamics is driven by the ME principle. One may dismiss this conclusion by arguing that this is merely a consequence of the second law of thermodynamics. However, one cannot talk about these folding trajectories without mapping the systems into information manifolds to begin with.
4. The Relative Importance of Tubulin Isotypes Unveiled by the Maximum Entropy Approach
4.1. Introduction
The ultimate goal of our work in the field of anti-cancer drug discovery is to investigate the relative importance of tubulin isotypes in eliciting a response of cancer cells to cytotoxic stress since tubulin is the target of some of the most successful anti-tumor drugs, such as taxanes and vinca alkaloids [34,35] used in cancer chemotherapy. In order to understand the complex behavior of various cancer cells exposed to a novel family of tubulin-binding compounds created as derivatives of colchicine, we propose to apply the ME approach to predict the expression levels of specific isotypes of tubulin in response to cytotoxic agents [15]. Six cancer cell lines as listed in Table 1, A549, MCF7, CEM, HeLa, M006X, and M010B, were used in this study and they were subjected to colchicine and 20 of its novel derivatives with significantly different binding affinities for each tubulin isotype, particularly, αβI, αβII, αβIII, and αβIV. Specifically, based on the assumption that cytotoxicity is correlated with drug affinity for the molecular target, we ask: “Given the information regarding the binding free energy between individual tubulin isotypes and colchicine derivatives and the IC50 values (effective concentration at which 50% of the cells are seen to experience drug-induced apoptosis) from cytotoxic measurements on the cell lines exposed to these drugs, what are the most likely expression levels of specific tubulin isotypes?”.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | Media |
|---|---|---|
| A549 | Human lung carcinoma | RPMI with 10% fetal bovine serum (FBS) |
| MCF-7 | Human mammary gland adenocarcinoma | RPMI with 10% FBS |
| CEM | Human T-lymphoblastoid from ALL | RPMI with 10% FBS |
| HeLa | Human cervical carcinoma | DMEM with 10% FBS |
| M006X | Human glioma cells | DMEM-F12 with 10% FBS and 1% Glutamine |
| M010B | Human glioma cells | DMEM-F12 with 10% FBS and 1% Glutamine |
4.2. Method
Again, this is exactly the type of question that the method of maximum entropy is designed to answer. The crux is then to determine the constraints relevant to tubulin isotype expression levels in cytotoxic measurements. The constraint equation that relates the cytotoxicity of colchicine derivatives given by the values of log IC50 and the binding free energy between tubulin isotype i and colchicine derivatives ΔGiα as:
where we use the superscript α = C or C' to denote the cell line exposed to colchicine or colchicine derivatives, respectively. Here, R = 8.314 J/K mol is the gas constant, T is the absolute temperature and M is the total number of tubulin isotypes. The ME method gives the probability distribution that is updated from a prior distribution with the information given by Equation (9) as:
where the partition function and the Largrangian multiplier βα are determined from Equation (9).
However, for binding free energy calculations, the thermodynamic cycle perturbation approach was proposed to overcome the difficulties related to the computational complexity of the problem [36]. Therefore, it is the relative binding free energy of C and C' to the α-tubulin dimer (ABT), ΔΔGbind which is calculated. By taking this into account and supposing that the tubulin isotype expression level when a cell line is exposed to colchicine is considered as a prior and given by , we can rewrite Equation (10) as:
where gi = −βCΔGiC = log(ZCPieC). Therefore, we can determine the expression level of each tubulin isotype i in the cell lines exposed to colcichine-based derivatives through this equation.
4.3. Results and Discussion
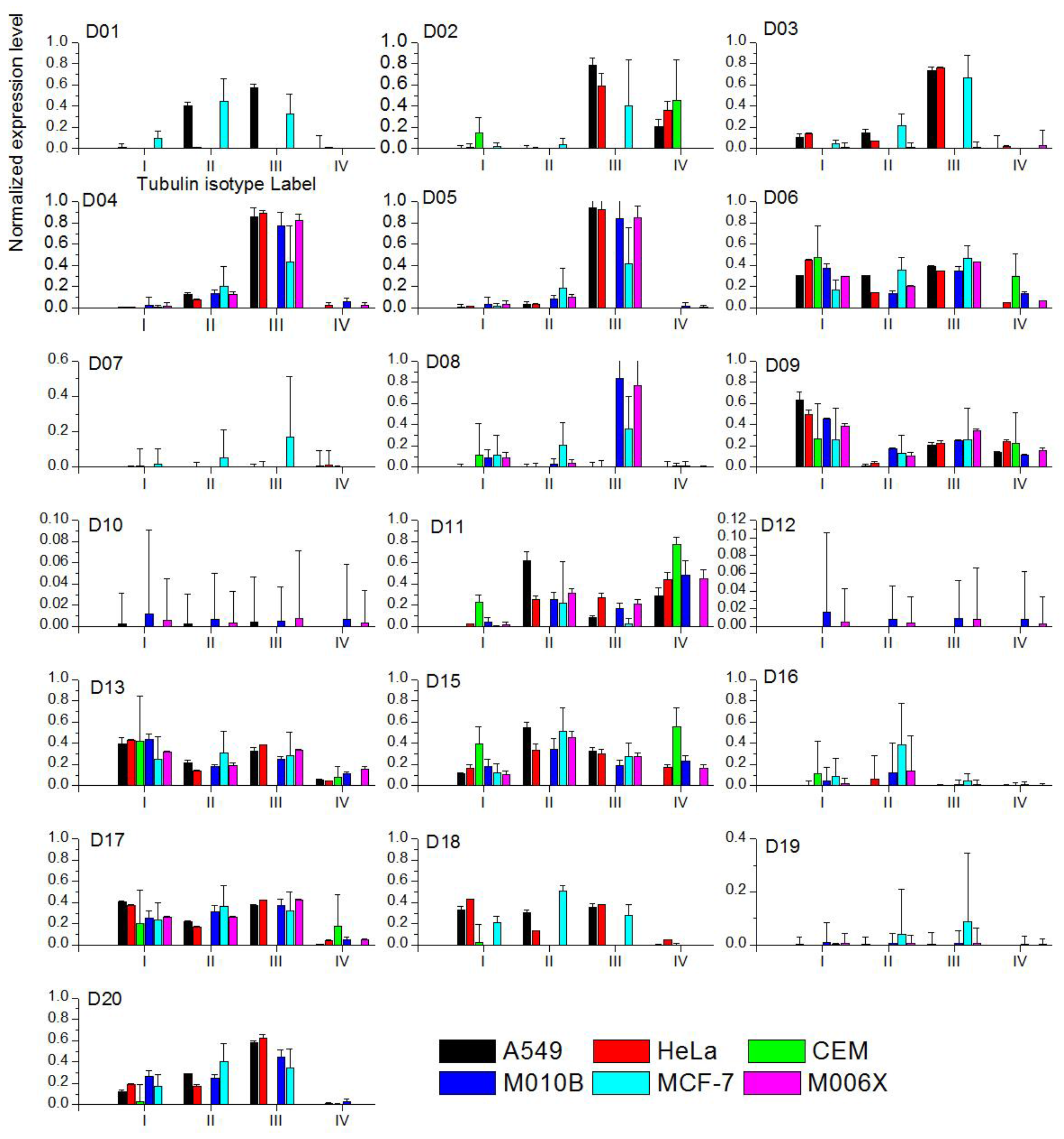
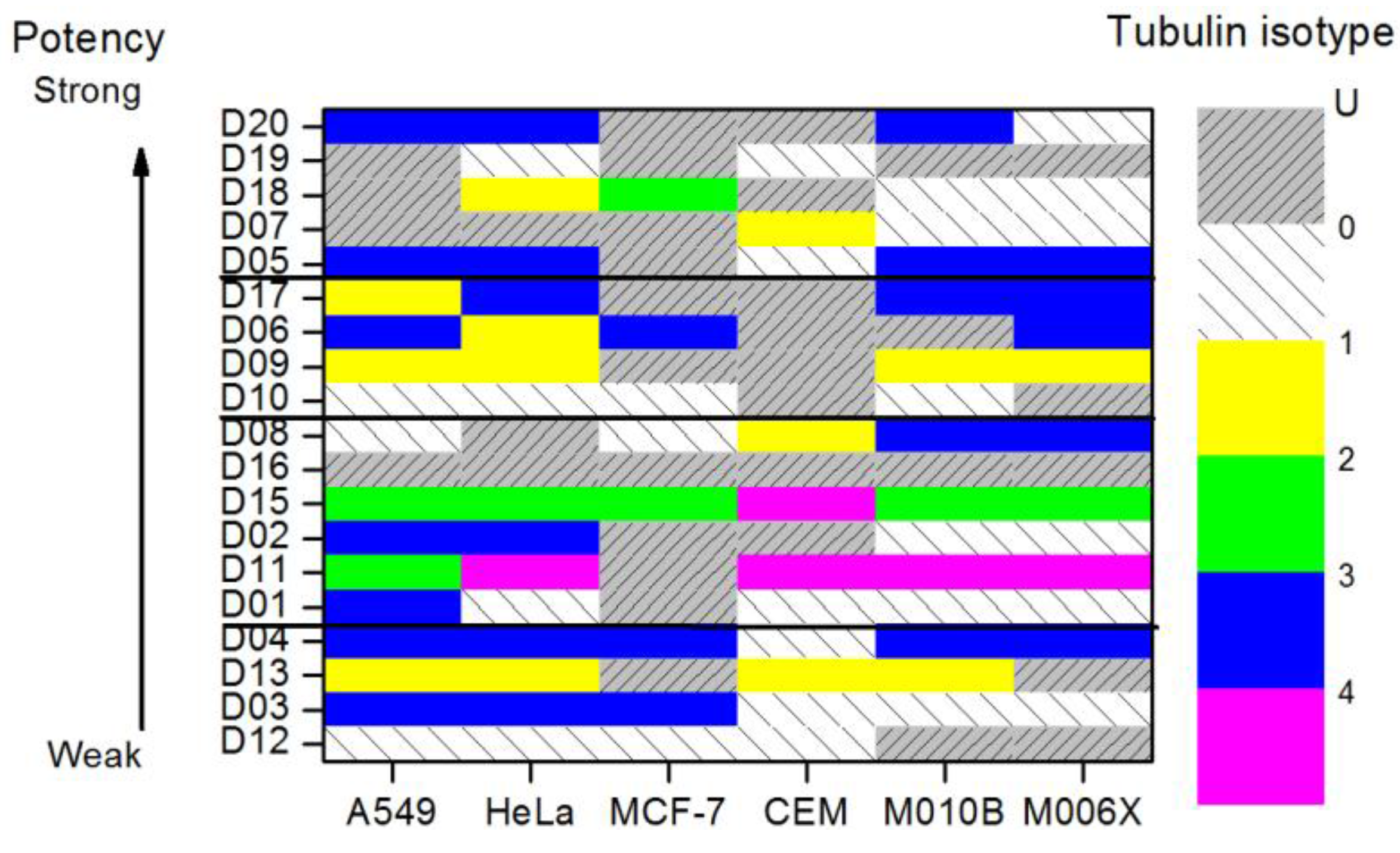
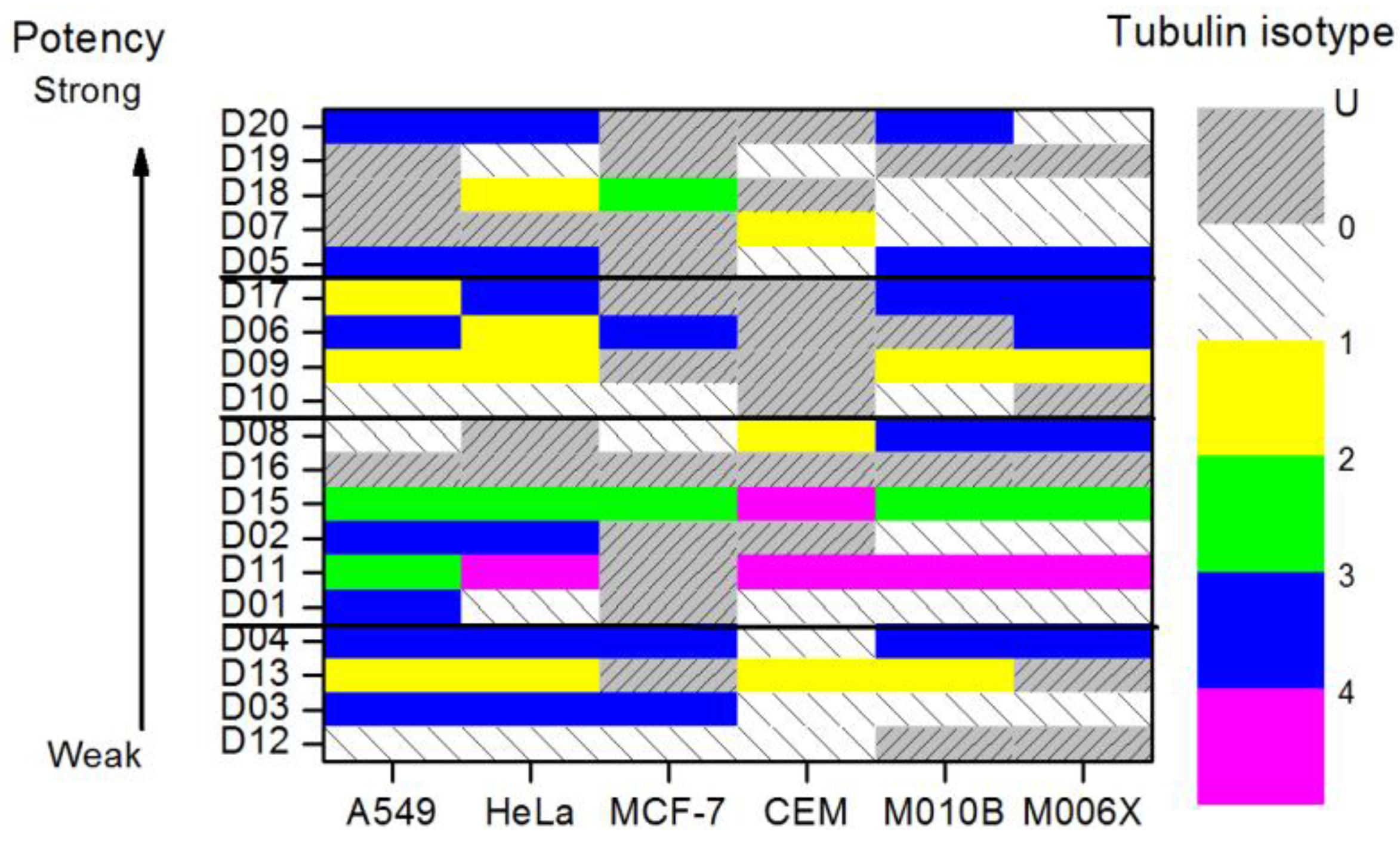
All six cell lines were studied given the tubulin isotypes expression levels in each cell line exposed to colchicine compounds as a prior. When the ME predicted expression level for a given colchicine derivative cannot be determined, zero expression levels are assigned. One can attribute it to the fact that relative binding free energy given such a derivative is ill defined. There is no analytical solution to Equation (9). The results of our numerical analysis are plotted in Figure 4. We further summarize and illustrate the tubulin isotype distribution with the highest expression levels in cell lines exposed to the twenty colchicine derivatives in Figure 5 to investigate the effects of the colchicine derivatives on the expression levels of tubulin. Note that the colchicine derivatives are plotted in the order of potency from weak at the bottom toward strong at the top based on the corresponding IC50 values. The remaining labels show the same order of tubulin isotype expression.
We have found by using our analysis that αβI and αβIII tubulin isoforms are the most important isoforms in establishing predictive response of cancer cell sensitivity to colchicine derivatives. However, since αβI tubulin is widely distributed in the human body, targeting it would lead to severe adverse side effects. Consequently, we have identified tubulin isotype αβIII as the most important molecular target for inhibition of microtubule polymerization and hence cancer cell cytotoxicity. Tubulin isotypes αβI and αβII are concluded to be secondary targets.
We have demonstrated the applicability of the maximum entropy approach in predicting cytotoxic effects based on limited information such as the relative binding energy values for the cytotoxic agents used. Specifically, given the relative binding free energy of tubulin isotype and colchicine derivatives, the proposed approach predicts the tubulin isotype expression levels in various cancer cell lines exposed to colchicine derivatives. Our studies also provide a better-defined molecular target for the action of these anti-mitotic drugs, namely, tubulin isotype αβIII, for optimized chemotherapy drug design as compared to earlier efforts in this area. By narrowing down the focus of tubulin targets to this isotype, which is most dramatically regulated by cancer cells when exposed to the colchicine derivatives, both the efficacy and specificity of treatment will hopefully be improved.
Figure 4.
The ME expression levels of four tubulin isotypes in six cell lines exposed to twenty colchicine derivatives. Color labels the cell lines. Note that D14 is removed since there is no sufficient data for the calculations.
Figure 4.
The ME expression levels of four tubulin isotypes in six cell lines exposed to twenty colchicine derivatives. Color labels the cell lines. Note that D14 is removed since there is no sufficient data for the calculations.

Figure 5.
The distribution of tubulin isotypes with the highest expression levels in six cell lines, where they are exposed to 20 colchicine derivatives. The color map level 1 to 5 indicates the types of tubulin isotypes. Label “U” denotes that there are multiple isotypes that have the same highest expression level. Label “0” denotes no ME prediction can be made. All the colchicine derivatives are sorted in the order of potency (logIC50 value).
Figure 5.
The distribution of tubulin isotypes with the highest expression levels in six cell lines, where they are exposed to 20 colchicine derivatives. The color map level 1 to 5 indicates the types of tubulin isotypes. Label “U” denotes that there are multiple isotypes that have the same highest expression level. Label “0” denotes no ME prediction can be made. All the colchicine derivatives are sorted in the order of potency (logIC50 value).
5. Entropic Fragment Based Aptamer Design
5.1. Introduction
In this last example, we show the role of entropy in de novo drug design. Particularly, we have developed an information-driven approach to design short DNA or RNA aptamer templates using the structural information of their intended targets [16]. Aptamers can bind to specific molecular targets including small molecules, proteins, nucleic acids, phospholipids and can also be targeted to complex structures such as cells, tissues, bacteria and other organisms. Because of their strong and specific binding through molecular recognition, aptamers are promising tools in molecular biology and have both therapeutic and diagnostic clinical applications [37,38,39,40]. They are designed through an experimental technique known as the Systematic Evolution of Ligands by EXponential enrichment (SELEX) [37,39]. SELEX consists of a number of rounds in which a RNA or DNA pool is incubated with the binding target and the non-binding or loosely binding sequences are discarded. The binding sequences are then amplified by polymerase chain reaction (PCR) to provide a pool of sequences for the next round. In practice, multiple rounds of selection and expansion are required before unique tightly binding sequences can be identified. Additionally, isolated aptamers will often need to be ref-engineered to reduce their sequence length and impart additional favorable biological properties. In the past, there were two general directions to take in developing in silico approaches. The first direction focuses on developing a computational approach to design structured random pools such as RagPools [41,42]. The second direction is to apply a virtual screening process [43]. However, several issues including the design of a screening library with reasonably good starting sequences and structural prediction tools to arrive at candidate structures and the associated structural distributions hinder the applicability of these approaches. We propose an information-driven theoretical approach to be free from these complications. This is expected to have a profoundly positive impact on aptamer design and increase the implementation of aptamer structures in various fields.
5.2. Theory
The concept of the proposed approach is to ask the question: “Given the structural information of the target, what is the preferred probability distribution of having an aptamer that is most likely to interact with the target?” instead of asking: “Given the structural information of the target and a customized library consisting of 1015 to 1016 different DNA or RNA sequences with the same initial lengths, what is the best binding mode?” in the virtual screening approach. Once we tackle the aptamer problem by asking the former question, the problem can be solved using information theory. By integrating the method of maximum entropy with the seed-and-grow strategy, we have proposed an entropic fragment based approach (EFBA). The proposed approach consists of three steps: (1) determine the probability distribution of the preferred first nucleotide based on the total energy of single nucleotide-target complex; (2) given the probability distribution of the preferred nucleotide obtained in the previous step, determine the probability distribution of preferred neighboring nucleotides based on the total energy of the dimer-target complex. By repeating this same procedure, one can obtain the joint probability distribution of an L-mer nucleotide sequence; (3) apply the entropic criterion [44,45] to determine the preferred sequence length L (the reader is referred to [16] for further details).
5.3. Results and Discussions
We chose the phospholipid phosphatidylserine (PS) as our target to examine the validity of the proposed approach. This was dictated by our interest in apoptosis where PS externalization is an early indication that this process is operational in the cell [46], which may be indicative of cytotoxic effects of chemotherapy drugs. To our knowledge, no aptamer has been described yet that targets PS and thus there is no benchmark for computational comparisons. We have, however, developed a model system to assess binding to PS using liposome technology and can therefore directly test the binding behavior of computationally derived sequences. In addition, we conducted four in silico experiments to provide theoretical insights into the experimental results.
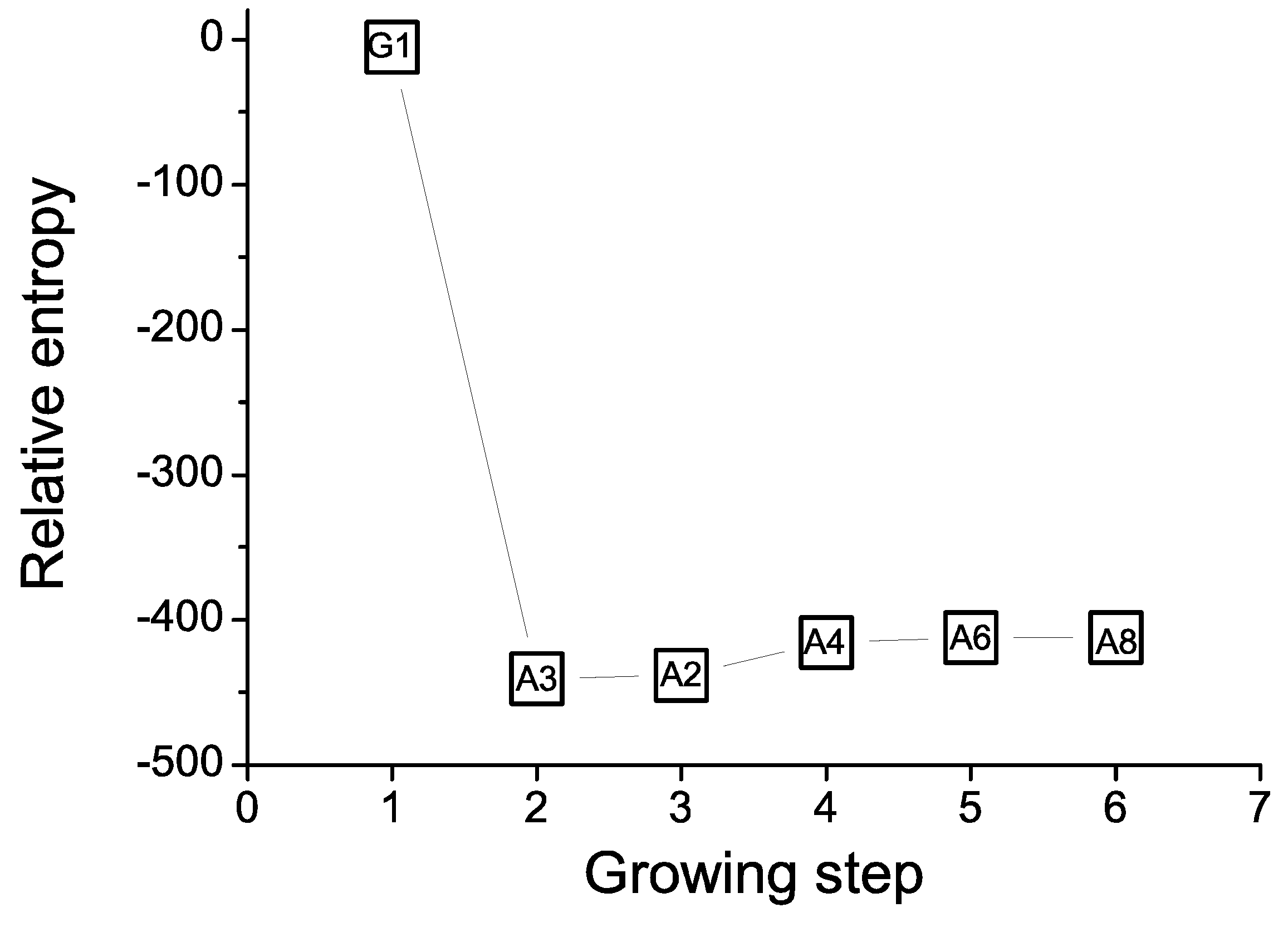
ME prediction. To design aptamers for PS, we utilize the total energy of PS and nucleotide fragments as information required for the design. However, since we are interested in studying apoptosis by investigating PS externalization, we have only considered the head group of PS for the design since the lipid portion of the molecule remains embedded in the cell lipid membrane. Figure 6 shows the relative entropy at each step of the growing process when the Largrangian multiplier is set to 1. The relative entropy value at each step indicates the effect of the corresponding nucleotide on the interactions of the complex when it is at a minimum. As a consequence, the portion grown in the aptamer after the step of saturation does not play an important role in the interactions and the preferred length L is the number of nucleotides grown before the step of reaching saturation. In this example, the saturation of relative entropy after the fifth step indicates that 6-mers (5'-AAAAGA-3', denoted by PS-aptamer I) are likely to be the best length for aptamers to interact with PS lipid head group. Two of the top four sequences, namely 5'-AAAAGA-3' (PS-aptamer I) and AAAGAC (PS-aptamer II), were selected for experimental assays and for in silico experiments intended as validation of the design method.
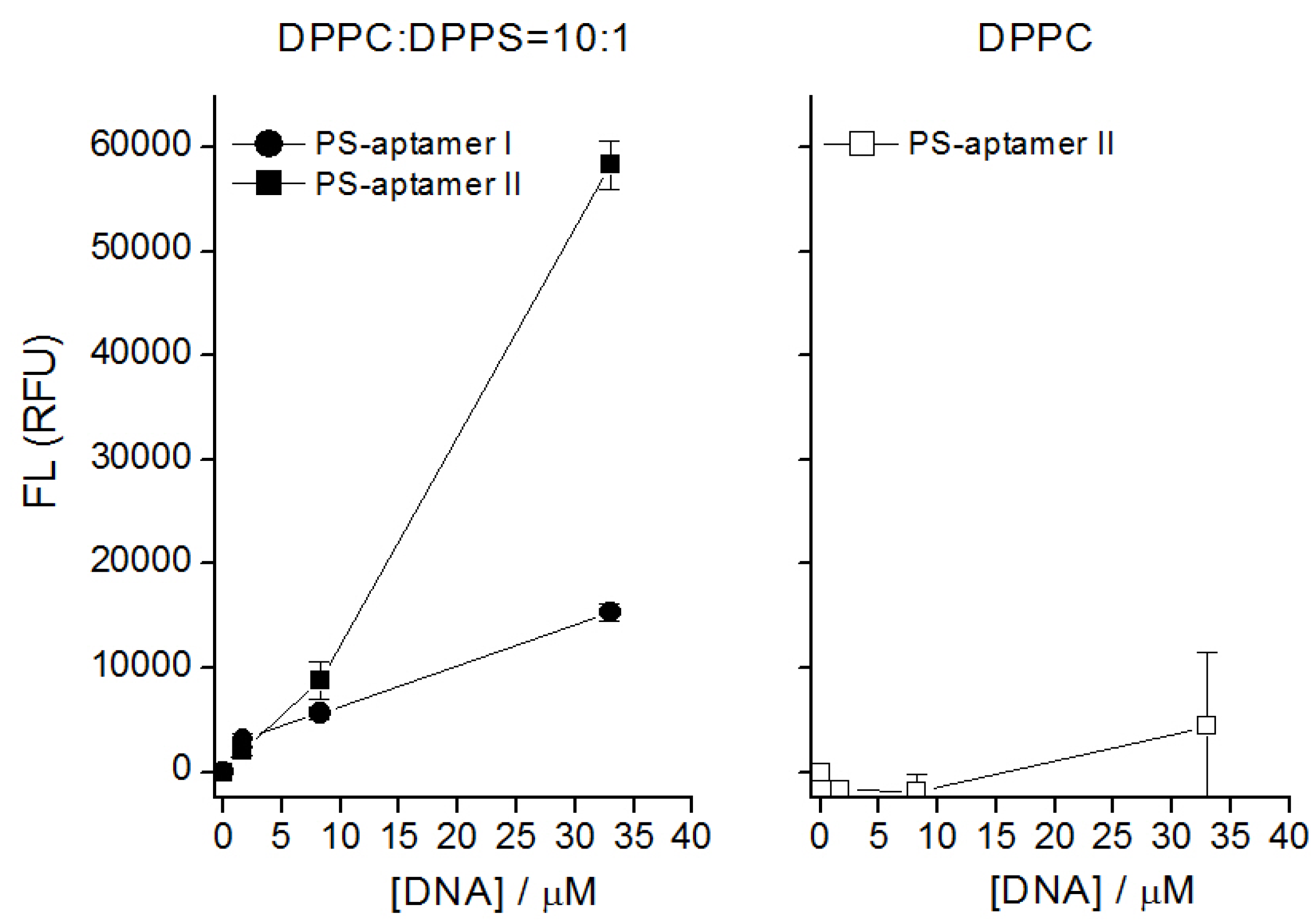
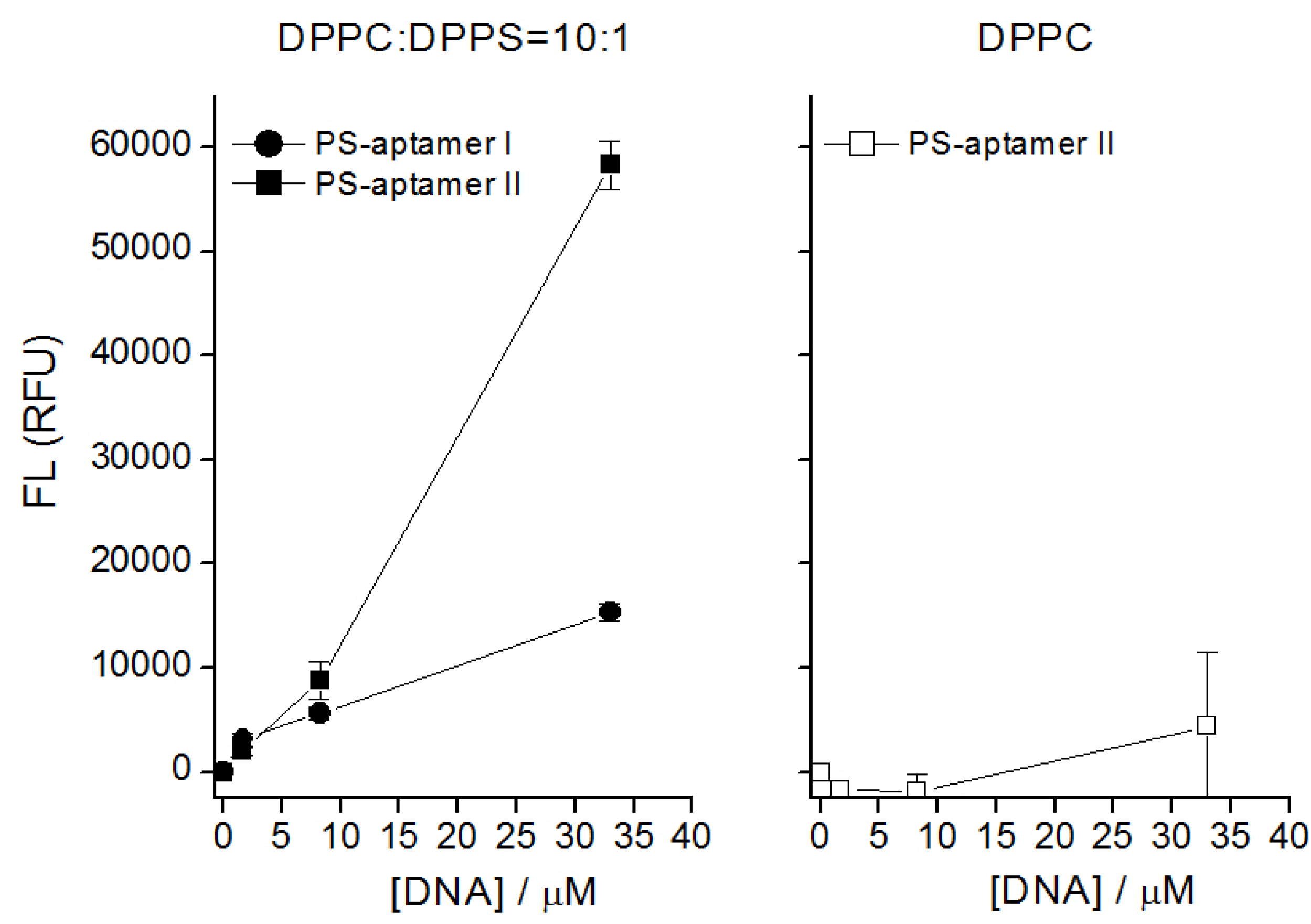
PS aptamer II binds specifically to PS in liposome assays and in simulations. Figure 7 indicates the fluorescence level from bound DNA aptamers using various concentrations of DNA aptamers with the two types of liposomes. We used mixtures of 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC) and cholesterol with 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DPPS). Two compositions were investigated DPPC:Chol:DPPS 10:5:1 and DPPC:Chol 10:5. The latter served as a control without PS available for binding. Two specific features are revealed here. First, both DNA aptamers bind with lipid liposomes. The poor or almost no binding with DPPC (see the right panel in the figure) suggests that the DNA aptamers bind specifically with DPPS (see the left panel in the figure). Thus, this indicates that the binding of the aptamers is specific to the lipid containing the serine headgroup. Second, although both DNA aptamers bind with DPPS, the results show PS-aptamer II has a higher fluorescence level than PS-aptamer I when the concentrations of DNA aptamers are increased beyond 0.165 nmol. This suggests that PS-aptamer II has a relatively stronger binding affinity than PS-aptamer I.
Figure 6.
The relative entropy of the calculation at each growing step for the case where β = 1 is shown. The figure shows that the relative entropy fluctuates around −400 after the 5th step. The relative entropy is shown along the Y-axis. The preferred nucleotide type at each step is labeled inside the open sqaure. The numbering beside each nucleotide denotes the connection end. For example, the phosphate at 5’ end of nucleotide A3 is connected with the oxygen atom at the 3’ end of G1 in second growing step. Then the oxygen atom at 3’ end of nucleotide A2 is connected with the phosphate at the 5’ end of G1 in third growing step. After the third step, the nucleotide with even numbering is connected with the phosphate of the 5’ end of the nucleotide with even numbering at previous growing step. The resulting sequence is 5’-A8-p-A6-p-A4-p-A2-p-G1-p-A3-3’ where p is phosphate.
Figure 6.
The relative entropy of the calculation at each growing step for the case where β = 1 is shown. The figure shows that the relative entropy fluctuates around −400 after the 5th step. The relative entropy is shown along the Y-axis. The preferred nucleotide type at each step is labeled inside the open sqaure. The numbering beside each nucleotide denotes the connection end. For example, the phosphate at 5’ end of nucleotide A3 is connected with the oxygen atom at the 3’ end of G1 in second growing step. Then the oxygen atom at 3’ end of nucleotide A2 is connected with the phosphate at the 5’ end of G1 in third growing step. After the third step, the nucleotide with even numbering is connected with the phosphate of the 5’ end of the nucleotide with even numbering at previous growing step. The resulting sequence is 5’-A8-p-A6-p-A4-p-A2-p-G1-p-A3-3’ where p is phosphate.
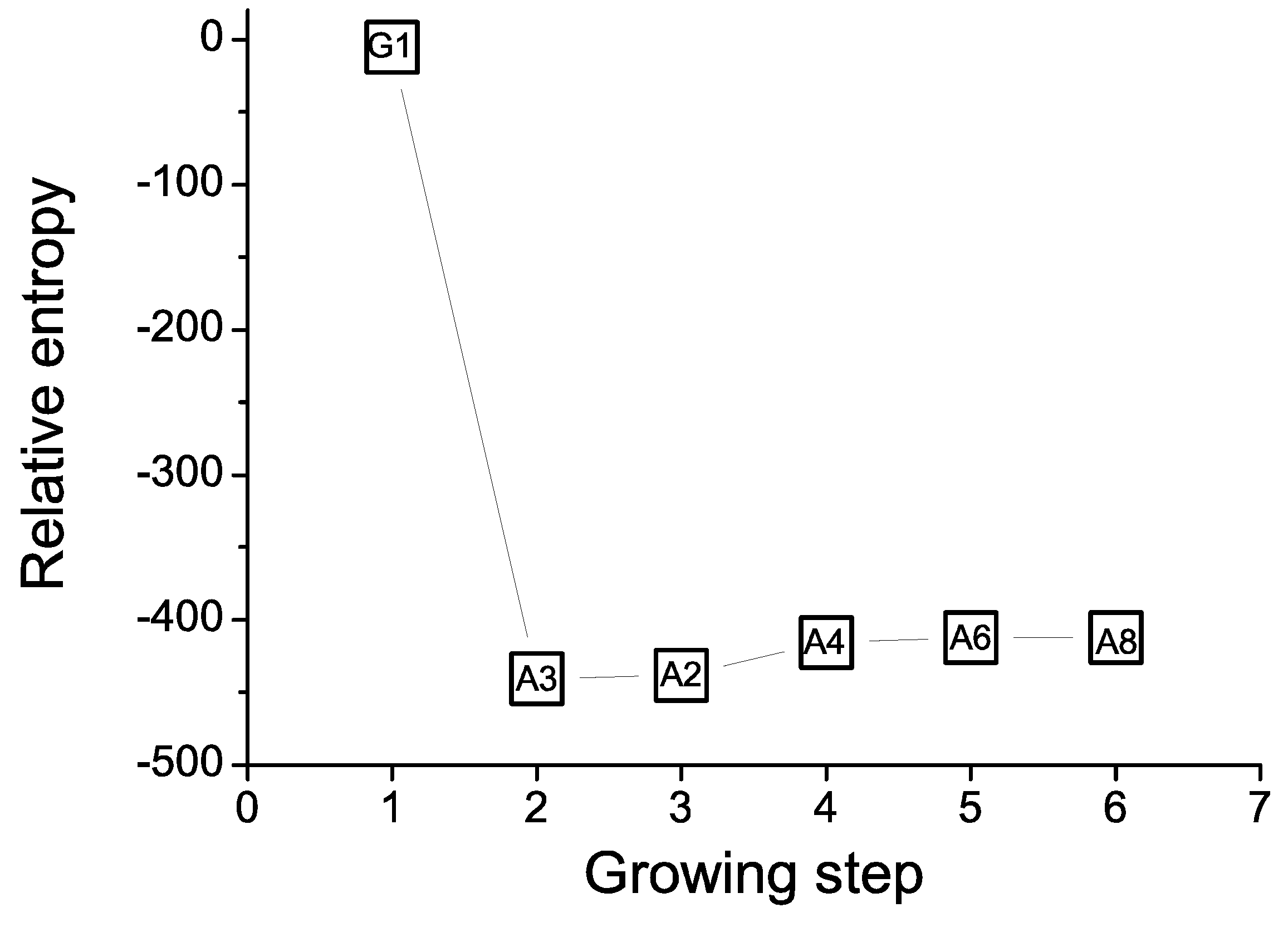
Figure 7.
Fluorescence (FL) measured in relative fluorescence units (RFU) versus aptamer concentration. Left panel: selective binding of two designed DNA aptamers with liposomes containing PS. Right panel: low non-specific binding of designed DNA aptamer with liposome containing only PC. DNA concentration shown here is the actual concentration used in lipids/cholesterol in HEPES buffer.
Figure 7.
Fluorescence (FL) measured in relative fluorescence units (RFU) versus aptamer concentration. Left panel: selective binding of two designed DNA aptamers with liposomes containing PS. Right panel: low non-specific binding of designed DNA aptamer with liposome containing only PC. DNA concentration shown here is the actual concentration used in lipids/cholesterol in HEPES buffer.
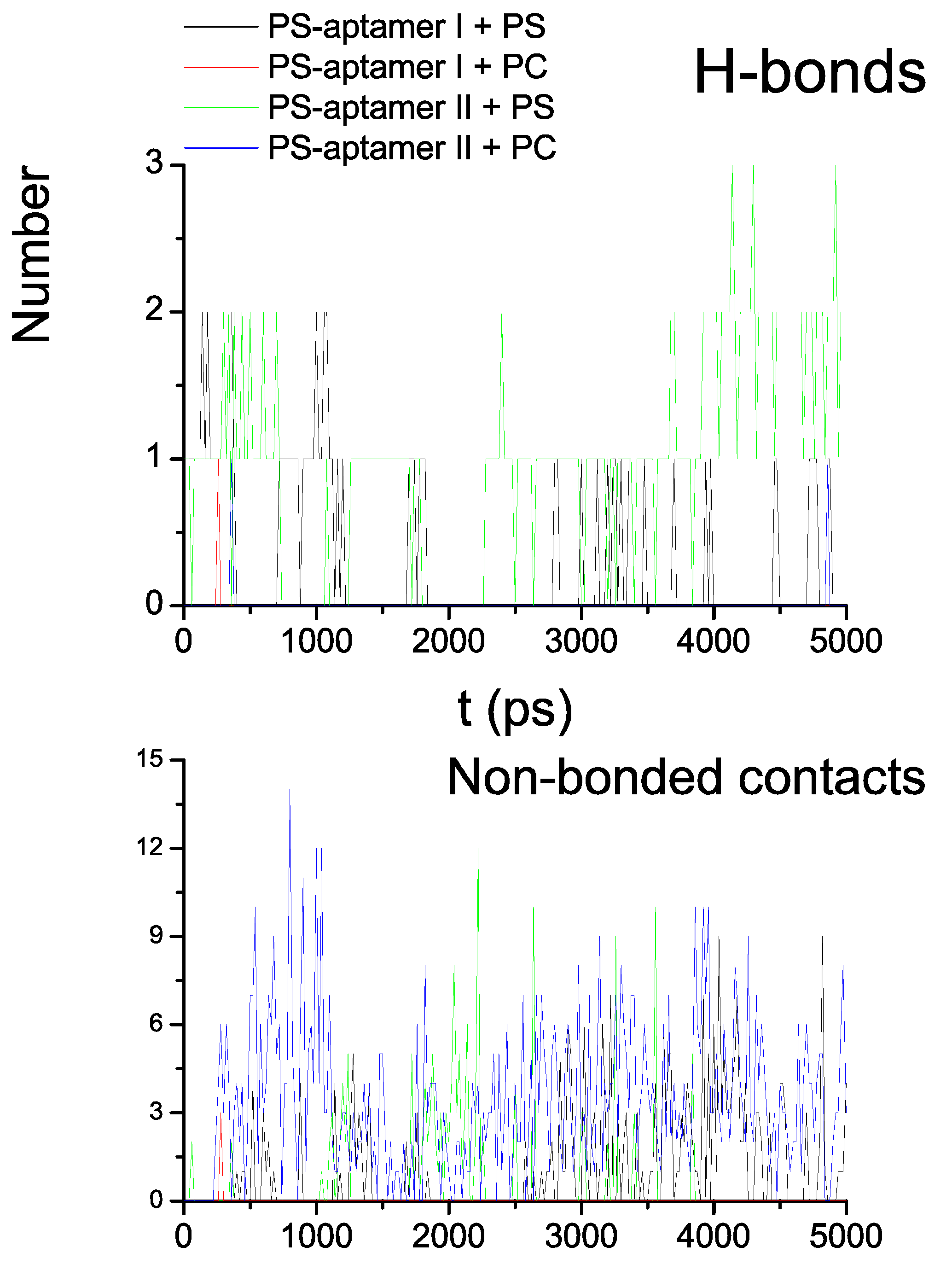
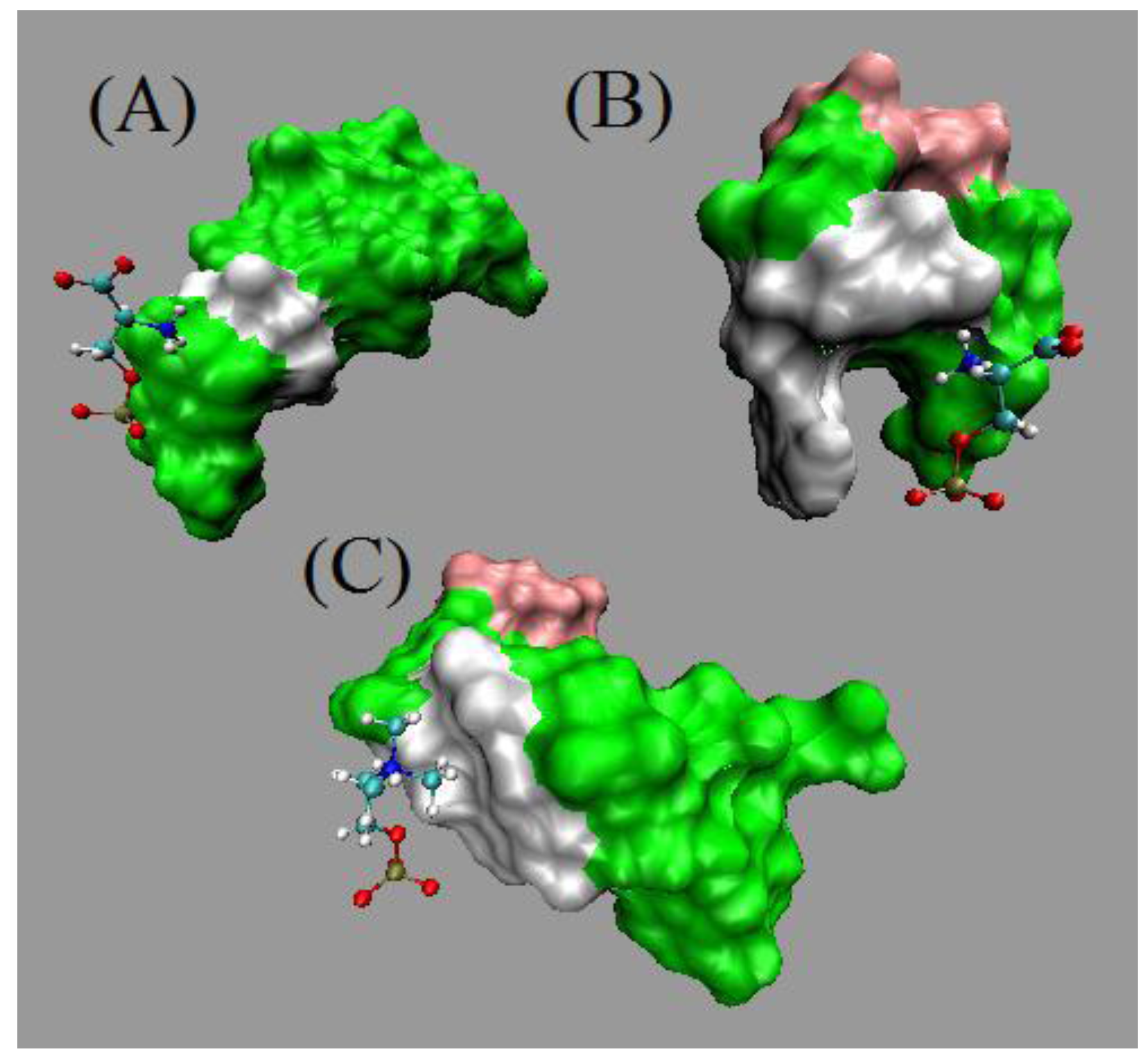
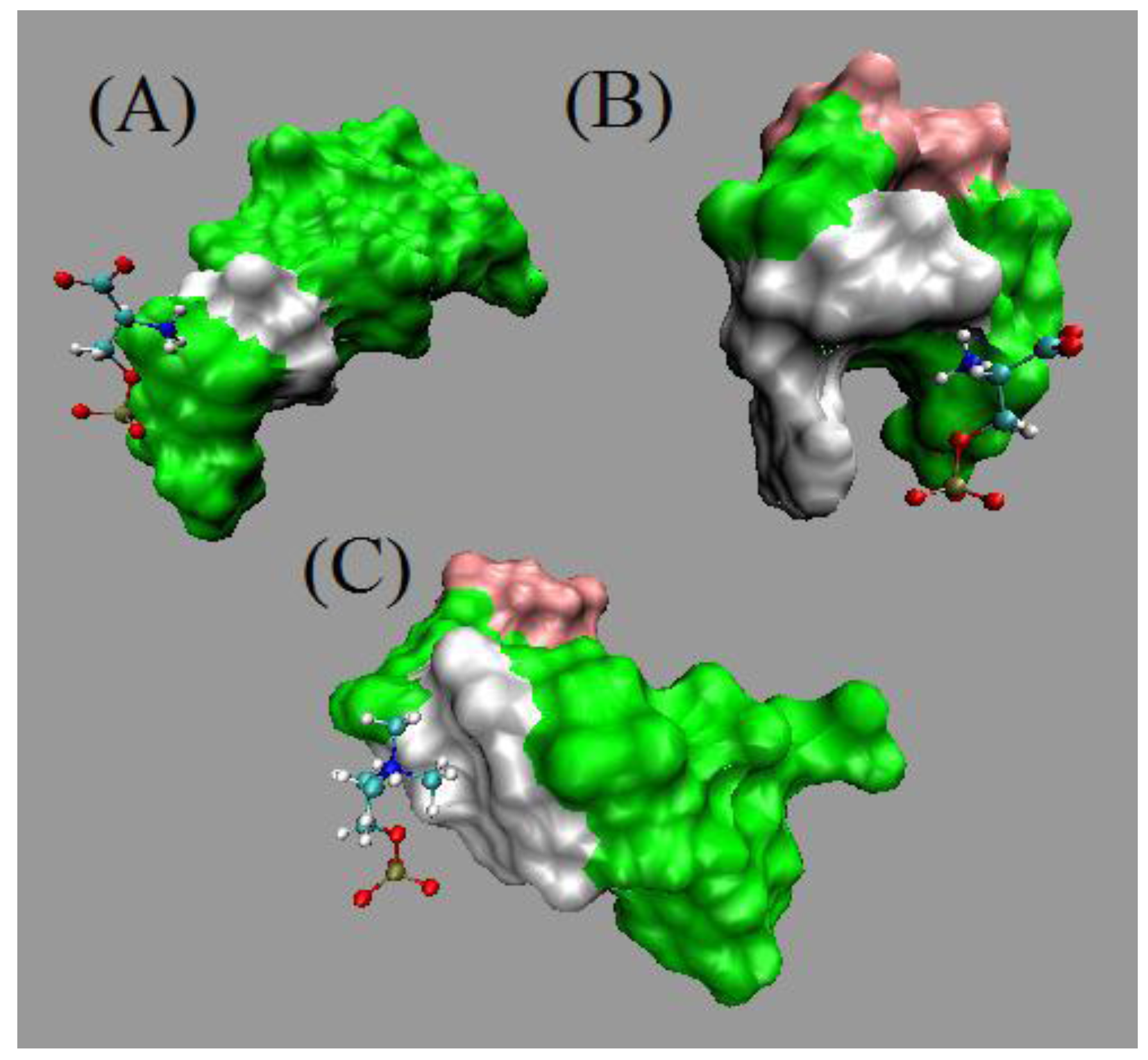
Based on in silico experiments, Figure 8 showed two types of interactions, hydrogen bond (H-bond) in the upper panel and non-bonded contacts in the lower panel identified during the four 5 ns MD simulations. The figure indicates that there are more hydrogen bonds constantly formed between PS and PS-aptamer II than between PS and PS-aptamer I after the 4th ns. Even though there are some non-bonded interactions between PS-aptamer I and PS formed, the PS-aptamer II is still considered to have stronger binding affinity than PS-aptamer I due to the hydrogen bond which provides a strong and direct interaction. Since no hydrogen bonds are observed between either PS-aptamer I or II and phosphatidylcholine (PC), which is another type of lipid that forms membranes, this suggests that both ME aptamers have stronger interactions with PS than with PC. Hence, the ME aptamers bind specifically to PS. The structures of the three complexes shown in Figure 9 at 5 ns highlight the binding modes of the ME aptamers. Panel (A) and (C) of Figure 9 show that only PS-aptamer I and II have close contacts with PS and PC, respectively. Panel (B), however, shows that PS-aptamer II forms a claw-shaped structure and PS is likely to be captured within the middle of the claw. Note that since PS-aptamer I drifts away from PC during simulations the corresponding structure is not presented in Figure 9. Furthermore, the ME aptamers are plotted using a surface representation, in which nucleotide A is represented by green, C by pink and G by green. Both results are not only consistent with our experimental results but also suggest a theoretical basis for the interactions of the ME aptamers with either PS or PC.
Figure 8.
The upper panel shows the number of hydrogen bonds formed between two ME aptamers and either PS or PC during simulations. The lower panel shows the number of non-bonded contacts.
Figure 8.
The upper panel shows the number of hydrogen bonds formed between two ME aptamers and either PS or PC during simulations. The lower panel shows the number of non-bonded contacts.
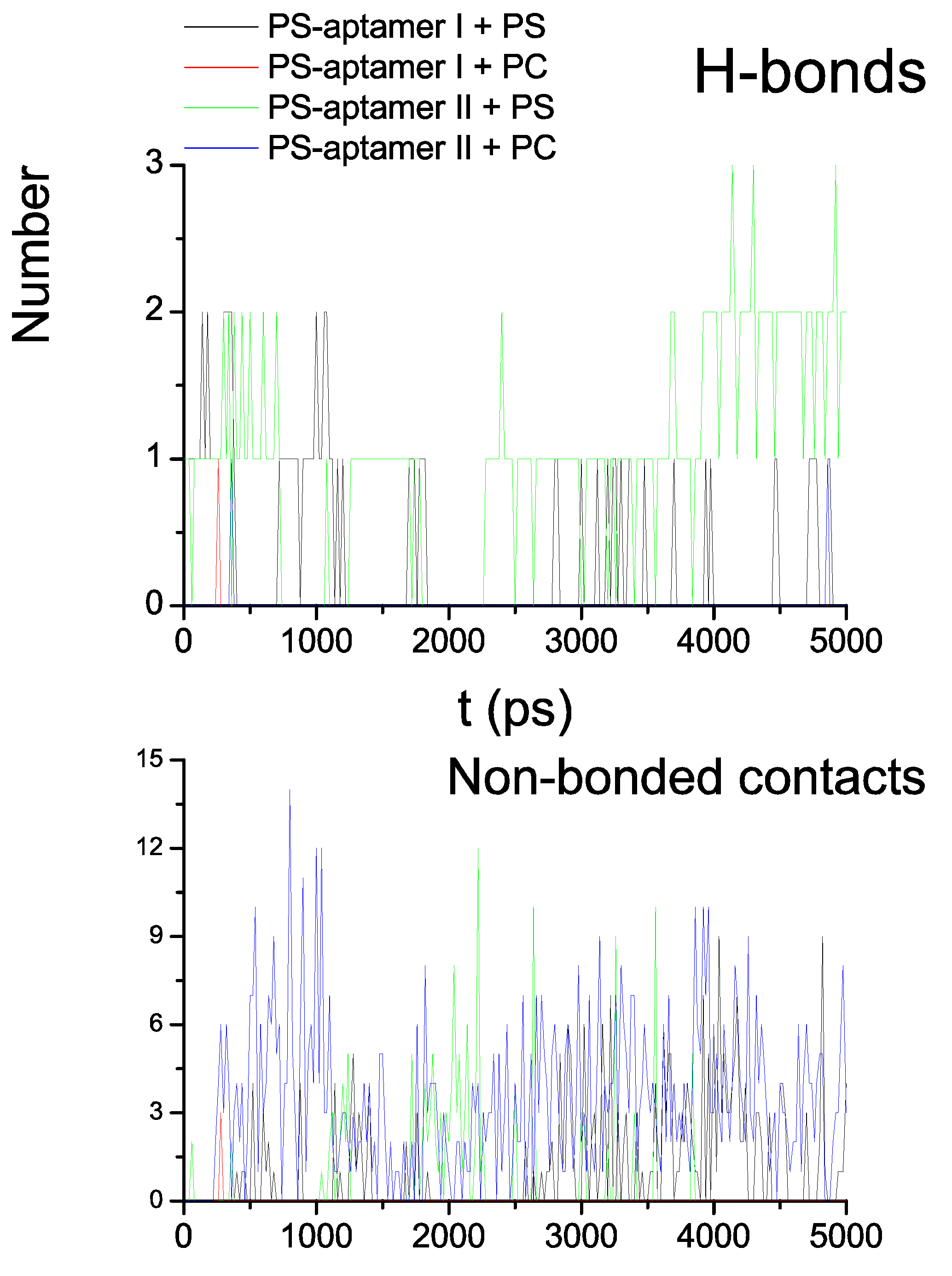
Figure 9.
The surface representations of the two ME aptamer structures at the 5th ns are presented, in which A is represented by green, C by pink and G by white. Panel (A) and (B) show the binding modes between two ME aptamers and PS, respectively. Panel (C) shows the binding mode between the PS-aptamer II and PC.
Figure 9.
The surface representations of the two ME aptamer structures at the 5th ns are presented, in which A is represented by green, C by pink and G by white. Panel (A) and (B) show the binding modes between two ME aptamers and PS, respectively. Panel (C) shows the binding mode between the PS-aptamer II and PC.
5.4. Comments
The EFBA is based on the method of maximum entropy and the structural information on the targets of interest. Since the method of maximum entropy is a universal and objective tool to process information with the least bias, one can expect the outcome to be preferred over others if sufficient information relevant to the problem posed is provided. In the case of designing PS aptamers, our computational and experimental results show a strong binding affinity and selectivity of the PS-aptamer II. It suggests that the PS-aptamer II is a potential imaging tool for detecting apoptosis in cancer cells subjected to chemotherapy treatments. This result is undergoing further investigation.
6. Conclusions
Traditional theoretical approaches to study biological systems have been developed based on laws of physics and chemistry. Although those approaches have shown great success, complicated many-body interactions still remain a challenge that often prevents one from obtaining exact solutions using these traditional approaches. Hence, one solution is to introduce appropriate approximations. The pioneering body work of Jaynes and Caticha suggests an alternative novel approach, which hinges on the use of entropy. The concept of entropy allows us to integrate laws of physics (energy and matter) and principles of inference (information) to formulate a comprehensive approach based on the statistical problems of interest such as those frequently encountered when quantifying biological effects.
In this article we have reviewed three distinctive biological examples that illustrate the role of entropy in various subjects stemming from fundamental issues arising in the applications involving drug design. The first example demonstrates that given REM-MD simulations, protein folding dynamics can be directly derived from principles of inference. The crux of the matter is to appropriately codify information relevant to the dynamics of many-body systems into an information manifold. The second example predicts tubulin isotype expression levels in cancer cell line studies when anti-cancer drugs, namely colchicine derivatives, are applied. This example is based on the integration of the method of ME, binding free energy estimates, and cytotoxicity measurements. By narrowing down the range of specific tubulin isotypes identified in this approach to those which are most dramatically regulated by cancer cells when exposed to the colchicine derivatives, both the efficacy and specificity of treatment will hopefully be improved. Such crucial causes of treatment failure as severe side effects can now be successfully mitigated with the information gained by our analysis. Finally, the third example presented shows a maximum entropy based approach for aptamer design. We have both experimentally and computationally shown that by providing the structural information concerning PS only, a PS aptamer obtained from this approach demonstrates a strong binding affinity and selectivity. This is a desired feature in the applications involving testing the efficacy of cancer chemotherapy.
Acknowledgements
This research was funded by the Alberta Cancer Foundation, the Allard Foundation, NSERC, the Canadian Breast Cancer Foundation and Alberta Advanced Education and Technology.
References
- Tseng, C.-Y.; Caticha, A. Using relative entropy to find optimal approximations: An application to simple fluids. Physica A 2008, 387, 6759–6770. [Google Scholar] [CrossRef]
- Dill, K.A. Theory for the folding and stability of globular proteins. Biochemistry 1985, 24, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.T.; Su, Z.Y.; Gwan, J.F.; Hao, B.L.; Hsieh, C.H.; Lee, H.C. Mean-field HP model, designability and alpha-helices in protein structures. Phys. Rev. Lett. 2000, 84, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J. Information, physics, quantum: The search for links. In Complexity, Entropy and the Physics of Information; Zurek, W.H., Ed.; Addison-Wesley: Redwood City, CA, USA, 1990; p. 1. [Google Scholar]
- Jaynes, E.T. Information theory and statistical mechanics. Phys. Rev. 1957, 106, 620–630. [Google Scholar] [CrossRef]
- Caticha, A. Entropic priors. In Bayesian Inference and Maximum Entropy Methods in Science and Engineering; Erickson, G., Zhai, Y., Eds.; AIP: Melville, NY, USA, 2004; AIP Conf. Proc. 707; p. 75. [Google Scholar]
- Shore, J.E.; Johnson, R.W. Axiomatic derivation of the principle of maximum entropy and the principle of minimum cross entropy. IEEE Trans. Inf. Theory 1980, IT-26, 26–37. [Google Scholar] [CrossRef]
- Shore, J.E.; Johnson, R.W. Properties of cross-entropy minimization. IEEE Trans. Inf. Theory 1981, IT-27, 472–482. [Google Scholar] [CrossRef]
- Giffin, A.; Caticha, A. Updating probabilities with data and moments. In Bayesian Inference and Maximum Entropy Methods in Science and Engineering; Knuth, K.H., Caticha, A., Center, J.L., Giffin, A., Rodríguez, C.C., Eds.; AIP: Melville, NY, USA, 2007; AIP Conf. Proc. 954; p. 74. [Google Scholar]
- Caticha, A. Maximum entropy, fluctuations and priors. In Maximum Entropy and Bayesian Methods in Science and Engineering; Mohammad-Djafari, A., Ed.; AIP: Melville. NY, USA, 2001; AIP Conf. Proc. 568; p. 72. [Google Scholar]
- Caticha, A. Entropic dynamics. In Maximum Entropy and Bayesian Methods in Science and Engineering; Fry, R.L., Ed.; AIP: Melville, NY, USA, 2002; AIP Conf Proc 617; p. 302. [Google Scholar]
- Caticha, A.; Cafaro, C. From information geometry to Newtonian dynamics. In Maximum Entropy and Bayesian Methods in Science and Engineering; Knuth, K.H., Caticha, A., Center, J.L., Giffin, A., Rodríguez, C.C., Eds.; AIP: Melville, NY, USA, 2007; AIP Conf. Proc. 954; p. 165. [Google Scholar]
- Caticha, A. From inference to physics. In Maximum Entropy and Bayesian Methods in Science and Engineering; de Souza Lauretto, M., de Bragança Pereira, C.A., Stern, J.M., Eds.; AIP: Melville, NY, USA, 2008; AIP Conf. Proc. 1073; p. 23. [Google Scholar]
- Tseng, C.-Y.; Yu, C.-P.; Lee, H.C. From laws of inference to protein folding dynamics. Phys. Rev. E 2010, 82, 0219141–0219149. [Google Scholar] [CrossRef]
- Tseng, C.-Y.; Mane, J.Y.; Winter, P.; Johnson, L.; Huzil, T.; Izbicka, E.; Luduena, R.F.; Tuszynski, J.A. Quantitative analysis of the effect of tubulin isotype expression on sensitivity of cancer cell lines to a set of novel colchicine derivatives. Mol. Canc. 2010, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-Y.; Ashrafuzzaman, M.D.; Mane, J.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. Entropic fragment based approach for aptamer design. Chem. Biol. Drug Des. 2010. submitted for publication. [Google Scholar] [CrossRef]
- Cox, R.T. Probability, frequency and reasonable expectation. Am. J. Phys. 1946, 14, 1–13. [Google Scholar] [CrossRef]
- Cox, R.T. The Algebra of Probable Inference; The Johns Hopkins Press: Baltimore, MD, US, 1961. [Google Scholar]
- Jaynes, E.T. Probability Theory: The Logic of Science; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Giffin, A. Maximum Entropy: The Universal Method for Inference. Ph.D. Thesis, The State University of New York at Albany, NY, USA, 2008. [Google Scholar]
- Amari, S.; Nagaoka, H. Methods of Information Geometry; American Mathematical Society: Providence, RI, USA, 2000. [Google Scholar]
- Jaynes, E.T. Information theory and statistical mechanics II. Phys. Rev. 1957, 108, 171–190. [Google Scholar] [CrossRef]
- Fisher, R.A. Theory of statistical estimation. Proc. Camb. Phil. Soc. 1925, 22, 700–725. [Google Scholar] [CrossRef]
- Rao, C.R. Information and accuracy attainable in the estimation of statistical parameters. Bull. Calcutta Math. Soc. 1945, 37, 81–91. [Google Scholar]
- Amari, S. Differential-Geometrical Methods in Statistics; Springer-Verlag: New York, NY, USA, 1985. [Google Scholar]
- Rhee, Y.M.; Pande, V.S. Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation. Biophys. J. 2003, 84, 775–786. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Han, J.; Kamber, M. Data Mining: Concept and Techniques, 2nd ed.; Morgan Kaufmann: San Francisco, CA, USA, 2006. [Google Scholar]
- Jaynes, E.T. Where do we stand on maximum entropy? In The Maximum Entropy Formalism; Levine, R.D., Tribus, M., Eds.; MIT Press: Cambridge, MA, USA, 1979; p. 15. [Google Scholar]
- Juraszek, J.; Bolhuis, P.G. Sampling the multiple folding mechanisms of Trp-cage in explicit solvent. Proc. Nat. Acad. Soc. U. S. A. 2006, 103, 15859–15864. [Google Scholar] [CrossRef] [PubMed]
- Dellago, C.; Bolhuis, P.G.; Csajka, F.S.; Chandler, D. Transition path sampling and the calculation of rate constants. J. Chem. Phys. 1998, 108, 1964–1977. [Google Scholar] [CrossRef]
- Bolhuis, P.G.; Chandler, D.; Dellago, C.; Geissler, P.I. Transition path sampling: Throwing ropes over rough mountain passes in the Dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. [Google Scholar] [CrossRef] [PubMed]
- Dellago, C.; Bolhuis, P.G.; Geissler, P.I. Transition Path Sampling. Adv. Chem. Phys. 2002, 123, 1–78. [Google Scholar]
- Owellen, R.J.; Owens, A.H.J.; Donigian, D.W. The binding of vincristine, vinblastine and colchicine to tubulin. Biochem. Biophys. Res. Commun. 1972, 47, 685–691. [Google Scholar] [CrossRef]
- Derry, W.B.; Wilson, L.; Khan, I.A.; Luduena, R.F.; Jordan, M.A. Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry 1997, 36, 3554–3562. [Google Scholar] [CrossRef] [PubMed]
- Tembe, B.; McCammon, J. Ligand-receptor interactions. Comput. Chem. 1984, 8, 281–283. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef] [PubMed]
- Hamula, C.L.A.; Guthrie, J.W.; Zhang, H.; Li, X.F.; Chris, L.X. Selection and analytical applications of aptamers. Trends Anal. Chem. 2006, 25, 681–691. [Google Scholar] [CrossRef]
- James, W. Aptamer. In Encyclopedia of Analytical Chemistry; Wiley& Sons Inc.: Hoboken, NJ, USA, 2000; pp. 4848–4871. [Google Scholar]
- James, W. Aptamers in the virologists’ toolkit. J. Gen. Virol. 2007, 88, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Gan, H.H.; Schlick, T. A computational proposal for designing structured RNA pools for in vitro selection of RNAs. RNA 2007, 13, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Shin, J.S.; Elmetwaly, S.; Gan, H.H.; Schlick, T. RagPools: RNA-As Graph-Pools-a web server for assisting the design of structured RNA pools for in vitro selection. Bioinformatics 2007, 23, 2959–2960. [Google Scholar] [CrossRef] [PubMed]
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucl. Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-Y. Entropic Criterion for Model Selection. Physica A 2006, 370, 530–538. [Google Scholar] [CrossRef]
- Chen, C.-C.; Tseng, C.-Y.; Dong, J.-J. New entropy-based method for variables selection and its application to the debris-flow hazard assessment. Eng. Geo. 2007, 94, 19–24. [Google Scholar] [CrossRef]
- Montaville, P.; Neumann, J.M.; Russo_Marie, F.; Ochsenbein, F. A new consensus sequence for phosphatidylserine recognition by annexins. J. Biol. Chem. 2002, 277, 24684–24693. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Tseng, C.-Y.; Tuszynski, J.A. Using Entropy Leads to a Better Understanding of Biological Systems. Entropy 2010, 12, 2450-2469. https://doi.org/10.3390/e12122450
AMA Style
Tseng C-Y, Tuszynski JA. Using Entropy Leads to a Better Understanding of Biological Systems. Entropy. 2010; 12(12):2450-2469. https://doi.org/10.3390/e12122450
Chicago/Turabian StyleTseng, Chih-Yuan, and Jack A. Tuszynski. 2010. "Using Entropy Leads to a Better Understanding of Biological Systems" Entropy 12, no. 12: 2450-2469. https://doi.org/10.3390/e12122450