Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? †

Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge 02139, MA, USA
*
Author to whom correspondence should be addressed.
†
Note added by the Publisher: The editors of the journal have been alerted to concerns over potential bias in opinions and bias in the choice of citation sources used in this article. We note that the authors stand by the content as published. Since the nature of the claims against the paper concern speculation and opinion, and not fraud or academic misconduct, the editors would like to issue an Expression of Concern to make readers aware that the approach to collating literature citations for this article was likely not systematic and may not reflect the spectrum of opinions on the issues covered by the article. Please refer to our policy regarding <a href=\\\\\\\\\\\\\\\'https://www.mdpi.com/about/controversial-articles\\\\\\\\\\\\\\\'>possibly controversial articles</a>.
Entropy 2012, 14(10), 1953-1977; https://doi.org/10.3390/e14101953
Submission received: 28 September 2012
/
Revised: 16 October 2012
/
Accepted: 16 October 2012
/
Published: 18 October 2012
(This article belongs to the Special Issue Biosemiotic Entropy: Disorder, Disease, and Mortality)
Abstract
:Autism is a brain disorder involving social, memory, and learning deficits, that normally develops prenatally or early in childhood. Frustratingly, many research dollars have as yet failed to identify the cause of autism. While twin concordance studies indicate a strong genetic component, the alarming rise in the incidence of autism in the last three decades suggests that environmental factors play a key role as well. This dichotomy can be easily explained if we invoke a heritable epigenetic effect as the primary factor. Researchers are just beginning to realize the huge significance of epigenetic effects taking place during gestation in influencing the phenotypical expression. Here, we propose the novel hypothesis that sulfates deficiency in both the mother and the child, brought on mainly by excess exposure to environmental toxins and inadequate sunlight exposure to the skin, leads to widespread hypomethylation in the fetal brain with devastating consequences. We show that many seemingly disparate observations regarding serum markers, neuronal pathologies, and nutritional deficiencies associated with autism can be integrated to support our hypothesis.
Keywords:
autism; epigenetics; cholesterol sulfate; DNA methylation; sulfotransferases; heparan sulfate; folate; cobalamin; zincPACS Codes:
87.19.xm; 87.19.xt; 87.19.xw; 87.18.Vf; 87.18.Sn; 87.19.lk; 87.19.lv; 87.19.um; 87.19.uj1. Introduction
Autism, and, more generally, autism spectrum disorder (ASD) is a developmental brain disorder involving social, cognitive, and memory deficits, whose incidence rates continue to climb unabated. The most recent estimate from the United States Centers for Disease Control now places the incidence rate in boys, who are affected far more often than girls, at one in 54 [1], with an overall rate among both genders of one in 88. Just being a twin, whether monozyotic or dizygotic, is a risk factor for autism [2]. This suggests that a nutritional deficiency in utero could be a factor, since twins would place twice the demand on any nutritional element. Thus, it supports the concept that epigenetic factors during gestation may be at play.
While many different markers have been observed to be abnormal in association with autism, reduced levels of plasma sulfate and subsequent reduced sulfation capacity are among the most consistent findings in autism research [3]. A recent interest in sulfated glycosaminoglycans (GAGS) has spurred investigations highlighting the importance of sulfate in the developing brain [4]. It is proposed here that sulfate depletion, caused by a combination of various environmental factors and genetic predispositions, is an important contributor to the development of ASD, and that epigenetics plays an important role in readjusting the phenotypical expression to cope with a sulfate deficiency.
The idea that impaired sulfate metabolism is a key factor in autism is not new [5,6,7]. An excellent review chapter [6] discusses several abnormal factors related to sulfate chemistry that have been found in association with autism. Observed serum imbalances include low plasma levels of free sulfate, abnormally high ratios of plasma cysteine to sulfate, and high loss of sulfite, sulfate and thiosulfate through the kidneys, as well as reduced levels of urinary thiocyanate [6]. Additionally, many sulfate-dependent neurodevelopmental processes have been found to be defective in association with autism. Neonatal development depends critically upon sulfate for myelination in the brain and for the development of perineuronal nets formed from chondroitin sulfate, which modulate GABAergic inhibitory signaling. Both the Purkinje cells in the cerebellum and the GABAergic cells in the hippocampus depend on chondroitin sulfate for ion exchange. The maturing of dendrites depends upon heparan sulfate proteoglycans, and impaired dendritic maturity is a characteristic feature of brains at autopsy for individuals with autism.
DNA methylation is increasingly recognized as an important regulatory mechanism during development [8]. Dramatic methylation changes take place during early embryonic development, and, in fact, methyltransferase-deficient mice produce nonviable embryos. Genome-wide demethylation takes place at the cavitation stage, and the reestablishment of appropriate tissue-specific methylation patterns takes place as the embryo matures. It is logical that insufficient supply of methyl donors would lead to hypomethylation of DNA, and that this would impact development in unpredictable ways.
It has been proposed that impaired methylation capacity during development may play a role in autism [9,10,11,12,13], and this is consistent with the observed deficiencies in the transsulfuration pathways, which are intimately connected with the methylation pathways, through the common substrate, methionine, an essential amino acid [14]. In [13], on the basis of relevant biomarkers, a link is suggested between increased oxidative stress and impaired methylation capacity in autism. The early embryo has a critical need for methylation associated with selective gene silencing and activation during differentiation. It has recently been determined that methylation of the coding regions of DNA encodes activation whereas methylation of the promoter regions encodes deactivation [9]. Since methionine is an important source of methyl groups for methylation, deficiencies in methionine translate into reduced methylation capacity, and therefore impaired activation and deactivation of tissue-specific genes. Such deficiencies can arise directly from oxidative stress induced by exposure to environmental toxins, particularly in the context of a genetic predisposition [10].
Recent research on 80 autistic children, compared to 75 age-matched controls, demonstrated that children with autism had a serum ratio of S-adenosylmethione (SAM) to S-adenosylhomoycysteine (SAH) (SAM/SAH ratio) that was only 50% of that of the controls. A similar result was found in [12], where children with autism were found to have decreased blood levels of glutathione, glutathione peroxidase, methionine, and cysteine, as well as increased concentrations of oxidized glutathione (GSSG). Significantly, in [15], it was shown that even the parents of autistic children exhibit a metabolic profile reflecting reduced methylation capacity, associated with elevated homocysteine levels. Since homocysteine readily crosses the placental membrane [16], and since excess homocysteine is associated with DNA hypomethylation, likely due to inhibition of DNA methyltransferase by SAH [17,18,19], impaired methylation capacity in the mother would likely translate into impaired methylation capacity in the fetus.
Different tissue types are associated with a unique signature of hypermethylated genes, leading to active gene expression of the proteins specifically needed by the particular tissue type. Recent research has begun to characterize the “methylome,” at least with respect to a small number of cell types. Particularly salient for our purposes is the functional subset of genes that are preferentially expressed through hypermethylation in neurons [9].
Using hidden-Markov model (HMM) techniques to label partially methylated regions and distinguish these regions as highly methylated in neurons, these authors were able to genomically map a set of genes with distinct neuronal functions. They established that genes associated with cell adhesion, ion transport, cell-cell signaling, synaptic transmission, neuronal differentiation and nerve impulse transmission were differentially activated in neurons, when compared to lung fibroblasts and embryonic stem cells. Insufficient activation of these genes through impaired methylation would be expected to cause pathological neuronal expression as is observed in autism.
In the remainder of this paper, we will first discuss the importance of sulfation in biology. We will then look specifically at the role of sulfate in the developing brain, focusing on serotonin and the glycosaminoglycans. We next turn our attention towards evidence of abnormal sulfate metablism in association with autism, including impaired sulfoconjugation capacity and increased sulfate excretion. We will then discuss available theories concerning possible causes of this impairment, including genetic effects and environmental effects. In particular, we will emphasize the effect of insufficient sunlight exposure to the skin and will argue that this issue goes beyond vitamin D3 deficiency. We further discuss how other known characteristics of children on the autism spectrum, such as gastrointestinal problems, increased androgen levels, and increased inflammation, can be explained by the sulfate deficit, and we will show that treatments aimed at repairing sulfate metabolism have shown some benefit. Finally, we will discuss the evidence for an epigenetic effect related to genomic hypomethylation in the fetus in association with autism. We conclude with a discussion section where we elaborate on our hypothesis and demonstrate how it can explain all of the complex factors associated with autism. Finally, we discuss simple lifestyle changes that could be implemented to address the problem.
2. The Importance of Sulfation
Many diverse molecules throughout the body require sulfation. Some examples include xenobiotics, which are inactivated and made water soluble through sulfation (this facilitates their excretion); neurosteroids, hormones, and catchecholamines, which are also inactivated by sulfation (this can regulate the local expression of their active forms); and glycosaminoglycans, polysaccharide chains that are sulfated in a highly unique pattern, which serves to modulate the structure and function of the proteoglycans that make up the extracellular matrix.
The universal sulfate donor for each of these reactions is 3’-phosphoadenosine 5’-phosphosulfate (PAPS). Synthesis of PAPS is the limiting factor in sulfation and involves a two-step reaction using inorganic sulfate as substrate, which consumes two ATP [20] molecules. PAPS availability is thus dependent on the availability of sulfate and ATP, and on the subsequent activity of two enzymes: ATP-sulfurylase and APS-kinase. Factors which can decrease PAPS availability, thereby reducing the sulfation of biological molecules, include a low-sulfur diet and administration of xenobiotics such as acetaminophen which are processed for elimination through sulfation [20].
Along with PAPS, the process of sulfation requires a sulfotransferase (SULT) enzyme which is specific to the substrate [21]. There are five SULT families, each family targeting a different set of substrates. The various sulfotransferases are expressed in many tissues throughout the body, including the brain. SULT1 isoforms have been identified in the fetal brain, and it is clear that significant alterations in sulfotransferase activity take place during development, suggesting that sulfotransferases play a critical role in the development of the central nervous system.
SULT1, which includes five subtypes, is the best known of the sulfotransferase families. The SULT1A class, which has been found in platelets, liver, breast, and gastrointestinal tract, as well as the brain [21], targets phenols and catchecholamines, while the SULT1B class targets thyroid hormones, the SULT1C’s target xenobiotics, and SULT1E’s target estrogenic steroids [22]. The SULT2 enzymes sulfonate cholesterol and the derived hydroxysteroids, such as pregnenolone, DHEA, and other neurosteroids. SULT3 catalyzes the formation of sulfamates, while SULT4 and SULT5 have not been fully characterized.
The SULT1A subclass appears to be most relevant to autism. Indeed, a genetic link has been found between mutations in the SULT1A locus and autism [23,24,25]. Decreased expression of SULT1A in both platelets and the gastrointestinal tract has also been observed in individuals with ASD [7,21] along with increased levels of neurotransmitters and catechols, which are substrates for sulfation [26]. Since sulfate is the rate-limiting factor in the synthesis of PAPS as substrate for the sulfotransferase reactions, low sulfate levels and low SULT1A levels have the potential to invoke the same pathology. We hypothesize that decreased expression of SULT1A, possibly even through epigenetic mechanisms, may be protective in autism, by forcing potentially toxic metabolites to be processed via glucuronylation rather than sulfation [27], thus sparing sulfate from elimination, but increasing the likelihood of adverse reactions to environmental phenols.
3. Sulfate’s Role in the Developing Brain
Sulfate is a vital nutrient required for proper development, and as such, normally flows in abundant supply from the mother to the fetus. The developing child receives sulfate from the mother through the placental villi and later through colostrum, the mother’s first milk [28]. Especially during the third trimester, renal reabsorption of inorganic sulfate increases in the mother, increasing serum sulfate levels. This causes an increase in inorganic sulfate concentrations in the mother’s amniotic fluid and an enrichment of cholesterol sulfate in the placental villi. It has been hypothesized that sulfation of cholesterol, making it amphiphilic, allows it to penetrate the placental barrier [29]. It also then becomes a potential source of sulfate to the fetus. Cholesterol sulfate levels in the villi rise dramatically throughout pregnancy. The concentration of cholesterol sulfate increases from 3.93 to 18.35 to 23.75 pmol/mg dry mass over the course of the three trimesters, a six-fold increase from the first trimester to the third trimester [30]. During this same period, serum inorganic sulfate in the fetus is even higher than serum inorganic sulfate in the mother [20].
Within the brain, sulfate transporters are expressed most strongly in the cerebellum and the hippocampus, implying that these two regions carry out important processes requiring sulfate regulation [31]. Interestingly, both of these brain regions have been implicated in the pathogenesis of autism [32,33]. Sulfate may work in the brain through two major pathways: as an integral component of the extracellular matrix, and as a deactivator of many different neurotransmitters, including serotonin, dopamine, DHEA, and pregnenolone.
3.1. Sulfation of Glycosaminoglycans
One important use of sulfate in the body is to supply sulfate groups to the glycosaminoglycans (GAGS), sulfated sugar molecules on the cell surface that have recently been investigated as a crucial component in a number of processes, including cell-cell signaling, fibroblast growth factor (FGF) regulation, axon growth and guidance, and brain development [34,35,36]. In particular heparan sulfate progeoglycans (HSPGs) are essential for neurite outgrowth and connectivity between neurons and their target cells [35], and have been shown to be important in neurogenesis, axonal guidance, and synapse development [36]. HSPGs appeared very early in development in the animal kingdom, predating multicellular organisms and appearing simultaneously with the first nervous systems [37].
Of special interest is a recent discovery that a postnatally induced heparan sulfate deficiency in the brain is capable of causing autistic behavior in mice, including impairments in social interaction, expression of stereotyped, repetitive behavior, and impairments in ultrasonic vocalization, all without causing detectable changes in cellular arrangement in the brain [38]. A deficiency in sulfate will cause a reduced supply of PAPS, which has been shown to reduce the sulfation of GAGS [20,39]. Since the sulfation of GAGS is very specific and highly regulated, reduced sulfation of these important molecules has the potential to cause serious problems.
3.2. Sulfation of Serotonin
A reduced supply of sulfate would likely impair the sulfation of serotonin, thus impairing its inactivation. Indeed, increased levels of serotonin were identified in 20–48% of children diagnosed with autism and are one of the most consistently replicated biochemical findings in autism [3,40,41] Not only do increased levels of serotonin affect day-to-day brain functioning, but they also affect the development and outgrowth of serotonergic neurons, with increased levels of serotonin causing a loss of serotonin terminals in rat pups [42]. Increased serotonin during prenatal brain development has been shown to cause autistic symptoms in rats, including decreased social bonding, sensory hyper-responsiveness, seizures and motor changes, and a loss of oxytocin-containing cells in the hypothalamus [43]. An increased risk to seizures [44] and deficits in oxytocin processing [45] have both been found in association with autism.
Consistent with the idea that excess levels of serotonin are involved in the pathogenesis of autism is the fact that prenatal cocaine exposure, which increases serotonin, is heavily associated with autism at a rate of 11.4% [46]. A strong connection exists between autism and Smith-Lemli-Opitz Syndrome (SLOS), which is caused by a defect in the enzyme catalyzing the last step in cholesterol synthesis. Fifty three of those with SLOS develop autism [47], which might also be explained through serotonin, as cholesterol depletion has been shown to cause a loss of serotonin transporter (SERT) activity [48].
4. Findings of Abnormal Sulfate Metabolism in Autism
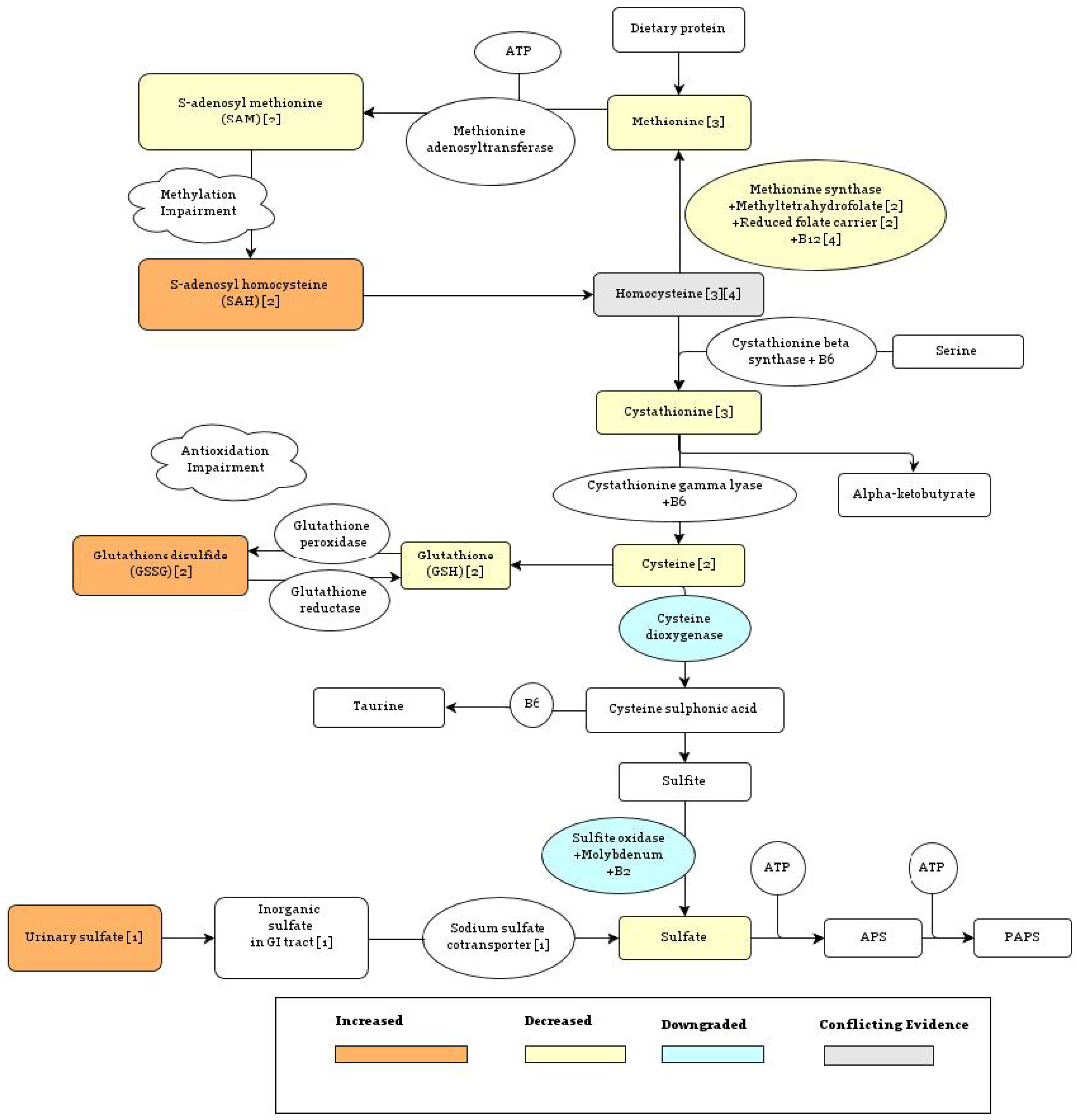
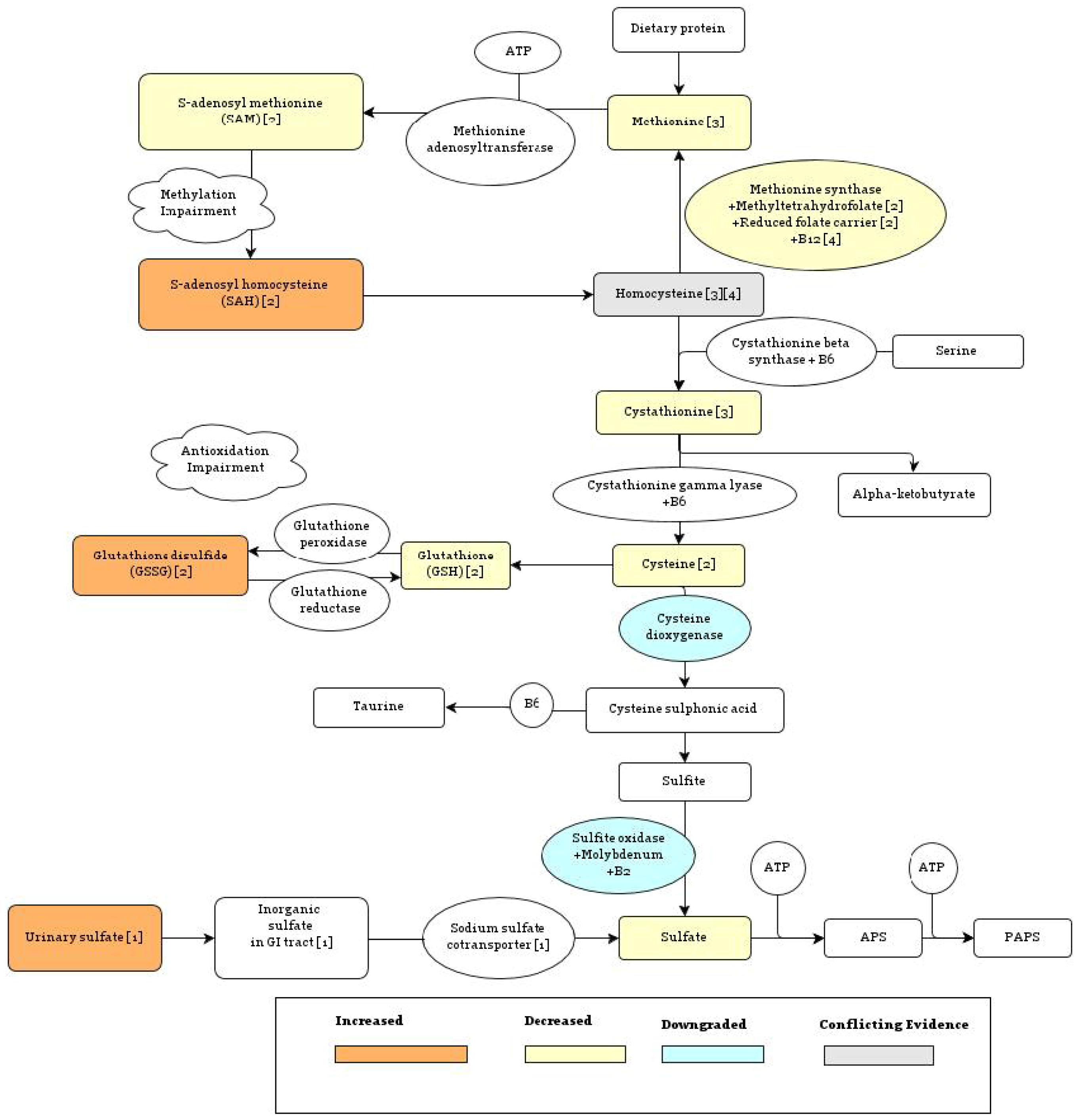
As summarized in Figure 1, individuals with ASD have significantly lower plasma levels of bound sulfate, free sulfate, and the sulfur metabolites cysteine, taurine, and glutathione (GSH), and significantly higher levels of oxidized glutathione disulfide (GSSG) relative to controls [49]. The role of homocysteine warrants further study, as it has been reported to be increased in autism [50] as well as decreased [51]. A decrease in homocysteine might reflect a generic deficiency in sulfur-containing amino acids, whereas an increase could reflect impairment in the pathway that regenerates methionine from homocysteine, which depends on both folate and cobalamin (vitamin B12) as cofactors (see Figure 1). Both free sulfate and reduced glutathione, which are depleted in autism, are important for detoxification, and lower levels of these metabolites are associated with oxidative stress, which could explain why inflammation biomarkers [44] and antioxidant biomarkers [5] tend to be higher in association with autism.
4.1. Evidence of Impaired Sulfoconjugation Capacity
Individuals diagnosed with ASD tend to have impaired sulfoconjugation capacity, which manifests as a difficulty in metabolizing various compounds, especially phenolic amines—dopamine, tyramine, and serotonin—which function as neurotransmitters [52,53], and which are inactivated through sulfation by SULT1A, as previously discussed.
4.2. Evidence of Increased Sulfate Excretion: Impairments in the Sodium Sulfate Cotransporter
In [49], it was found that children diagnosed with autism have on average serum sulfate levels that are reduced to 33% of the levels found in controls. These abnormally low levels of free sulfate can be explained in part by reduced resorption of inorganic sulfate in the kidneys. Normally, the level of sulfate in the body is tightly regulated by membrane transport proteins in the renal proximal tubule, which include the sodium sulfate cotransporter NaSi-1 [54]. However, children diagnosed with autism were found to have high urinary sulfate in addition to low plasma sulfate levels [7], suggesting that, in these individuals, the process of inorganic sulfate uptake and retention may have gone awry. Interestingly, the resorption of sulfate ions depends on the presence of sulfated proteins in the renal proximal tubule, so that, as sulfate levels decrease, the resorption of sulfate may decrease in a positive feedback loop [7].
{kind=link}
{kind=link}
5. Potential Causes of Abnormal Sulfate Metabolism
A decreased availability of sulfate can arise in the child during critical periods of brain development through a combination of genetic and environmental factors which can act prenatally (through the mother) or postnatally (directly on the child). The relationship between genetic mutations and autism is complex, but it has been proposed that an interaction between multiple susceptibility alleles and environmental factors could create a metabolic imbalance leading to autism [55], and we suggest that the common element among many of these genetic factors could be impaired sulfate metabolism.
5.1. Genetic Factors
While genetic links with autism remain relatively weak and elusive, there are several genotypic differences involving sulfate metabolism and heparan sulfate production which are related to autism. These include variants in the components necessary for the methionine synthase reaction (see Figure 1), sulfotransferase enzymes and molecules which show affinity for heparan sulfate.
5.1.1. Methionine Synthase Reaction
There are two routes for the metabolism of homocysteine in the body, the first of which leads to methionine through the methionine synthase reaction, and the second of which leads to cystathionine, catalyzed by cystathionine beta synthase (see Figure 1). The methionine synthase route enables methylation, while the cystathionine route protects from oxidation. Thus, low methionine levels will lead to reduced methylation and low cysathionine levels will lead to increased oxidative stress. Due to the fact that the methionine synthase reaction relies on B12, folate, and the reduced folate carrier, a lack of any one of these components will ultimately impair methylation. Zinc deficiency may also play a role.
Several genetic variants associated with the conversion of homocysteine to methionine have been associated positively with increased risk for autism, including the genes involved in the transport of folate and the transport of B12. Methionine synthase, a B12 dependent enzyme, catalyzes a methyl transfer from N5-methyltetrahydrofolate to homocysteine using a zinc ion to activate the homocysteine substrate [56]. This reaction is also dependent on the oxidative state of its surroundings, with increased oxidation leading to decreased methionine synthase activity, directing production of homocysteine away from methionine and towards cystathionine [10]. This will help to alleviate the oxidative burden through the metabolism of cystathionine to reduced glutathione, as illustrated in Figure 2 [57], but will decrease the bioavailability of methyl groups.
In order for the methionine synthase reaction to occur, reduced folate must be present inside the cell. Reduced folates are taken up by cells in a tightly regulated process governed by the reduced folate carrier (RFC) [58]. The RFC1 G allele, which is associated with impaired cellular uptake of folate [59], is overrepresented in children with autism. When compared with the AA genotype, the RFC-1 GA or GG genotypes conferred twice the risk for autism [55].
Another gene involved in the transport of folate is methyltetrahydrofolate reductase (MTHFR). The MTHFR 677T allele, which is associated with reduced synthesis of metabolically active folate, also confers a higher susceptibility to autism, especially when paired with the RFC1 G allele. In fact, a combination of the RFC-1 80GG allele and the MTHFR 677CT genotype was found to increase the risk of autism three fold [55]. Overall, these data seem to support the idea that a genetic tendency toward a folate deficiency induced by impaired folate transport may alter methionine metabolism and cause a predisposition towards autism.
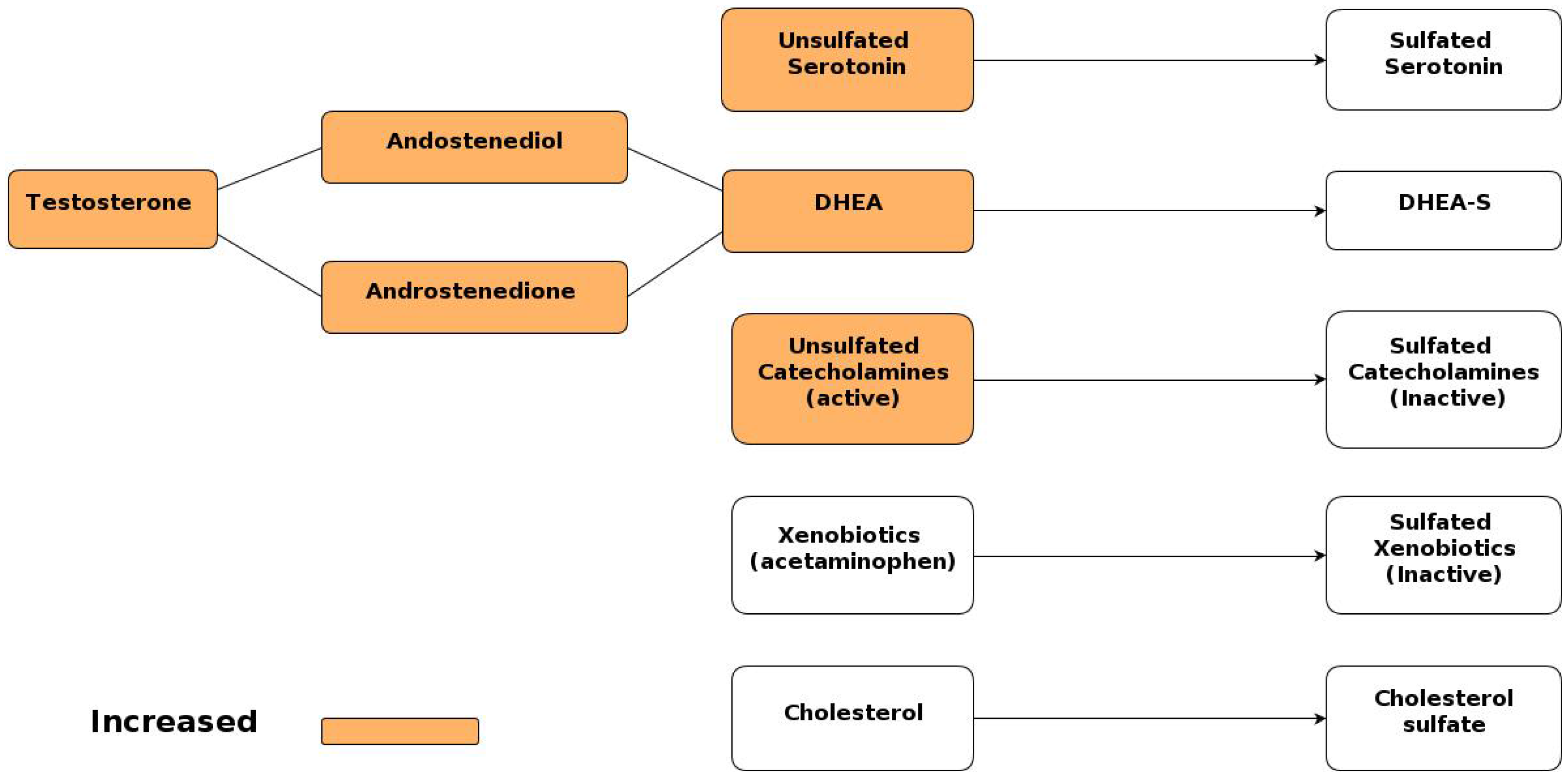
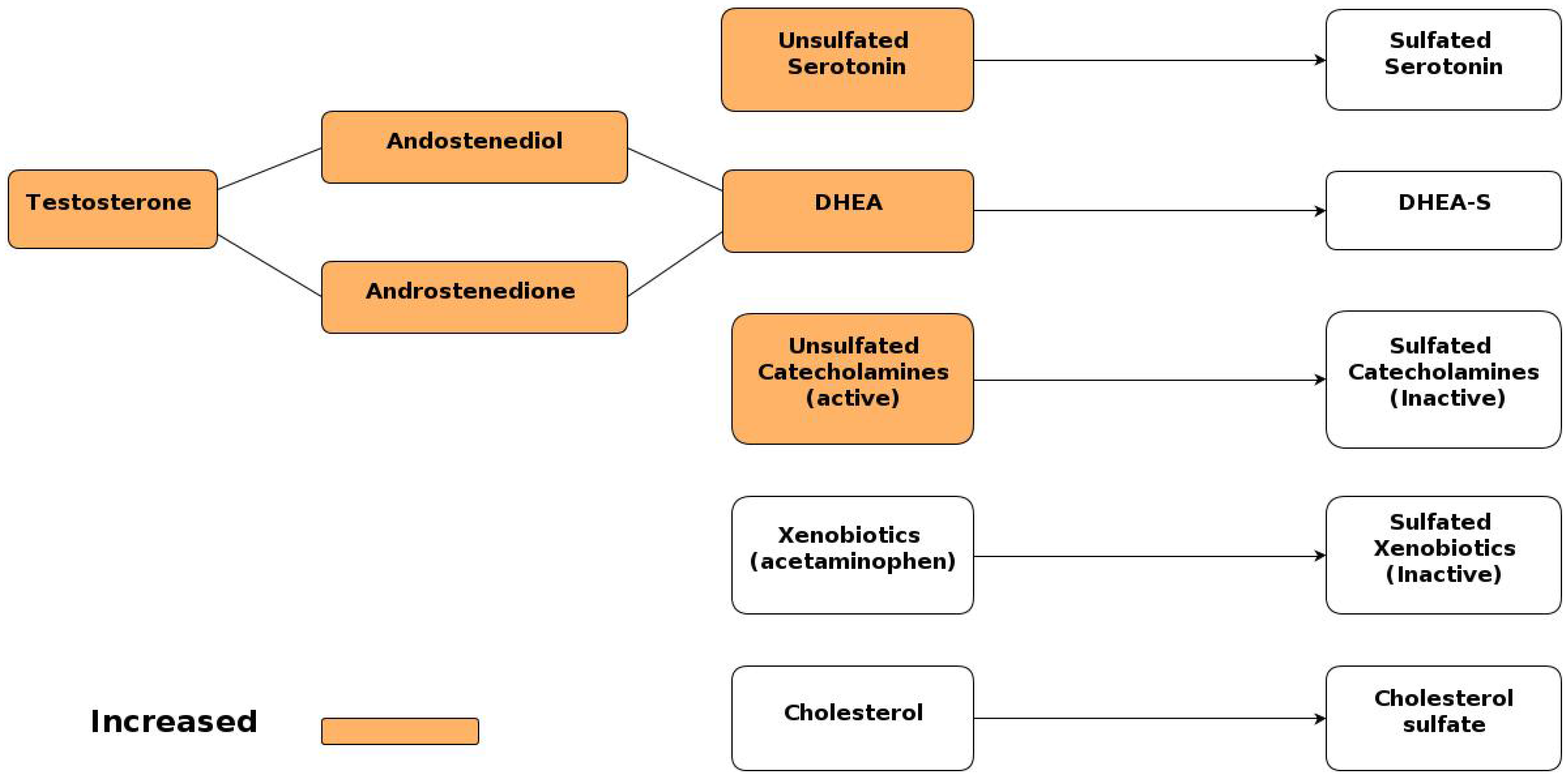
Figure 2.
Sulfoconjugation of important metabolites. Metabolites present in excess in autism are highlighted in orange.
Figure 2.
Sulfoconjugation of important metabolites. Metabolites present in excess in autism are highlighted in orange.
Interestingly, further studies report that a polymorphism in the reduced folate carrier (RFC) in the mother is even more strongly associated with autism than a polymorphism in the child [60]. In the same study, elevated homocysteine, adenosine, and S-adenosyl homocysteine were also noted as risk factors in the mother.
This suggests that the consequence of less effective methionine metabolism might be more relevant during the period when the fetus is developing in utero. Given that hyperhomocysteinemia, along with reduced levels of B12 and folate, is associated with obesity [61], this might help to explain the association between obesity in the mother and autism risk in the child [62].
Along with folate, cobalamin is also a critical cofactor in the methionine synthase reaction. One protein of interest in this process is transcobalamin II (TCN2), the transport protein which mediates the cellular uptake of B12 [63]. In this case, the TCN2776GG variant, which has a decreased binding affinity for B12, reduces its cellular uptake. This allele was found to increase the risk of autism 1.7-fold [55]. Indeed, it has been observed that B12 deficiency is associated with developmental regression that in many ways parallels the observed pathology associated with autism [64].
hydrolyzes homocysteine thiolactone [65], thus participating in its conversion to sulfate [66]. An association between reduced PON1 activity and autism in the United States was reported in [67]. Factors which might have contributed to this deficiency that were considered included excess dietary high fructose corn syrup (HFCS), insufficient dietary magnesium, and exposure to mercury. An association between PON1 deficiency and HFCS exposure has been confirmed in rats [68].
Overall, low methionine levels are observed in many children diagnosed with autism. As oxidative stress reduces methionine synthase activity [57], a combination of genetically impaired folate transport, B12 transport, and oxidative stress might work in tandem to produce this outcome.
5.1.2. Sulfotransferase Enzymes
Further along in the transsulfuration pathway, the sulfotransferase enzymes act to add sulfate groups to glycosaminoglycans, such as heparan sulfate, which among their many other roles are crucial components of neural development. Interestingly, two different sulfotransferase enzymes, phenol-sulfotransferase-P and heparan sulfate (glucosamine) 3-O-sulfotransferase 5 (HS2ST5), have been implicated in autism. In [53], it was found that all 40 children with autism who were tested showed decreased levels of the enzyme, phenolsulfotransferase-P (PST). Impaired PST function could lead to impaired elimination of the phenols found in food sources.
Finally, in a study done on two cohorts of European ancestry [69], a genetic association has been found between autism and a variant of the heparan sulfate (glucosamine) 3-O-sulfotransferase 5 (HS3ST5) gene. Heparan sulfate glucosamine 3-O-sulfotransferase catalyzes the transfer of a sulfate group from PAPS to [heparan sulfate]-glucosamine, forming [heparan sulfate]-glucosamine 3-sulfate, a critical component of the sulfated proteoglycans making up the extracellular matrix proteins. Impaired heparan sulfate supply is associated with autistic-like behaviors in mice [38].
5.1.3. Autism Associated Genes with Affinity for Heparan Sulfate
To further support the idea that a heparan sulfate deficiency in the brain is responsible for the onset of autism in some cases, two genes in particular, CNTNAP2 and NRXN1, which have been implicated in autism, encode proteins that have an affinity for heparan sulfate [70,71,72]. It could be that when there is a heparan sulfate deficiency, a less effective version of these two enzymes will decrease the efficacy of the heparan sulfate even further. Both CBTBAP2 (contactin associated protein-like 2) and NRXN1 (neurexin 1) are members of the neurexin family and act as cell adhesion molecules and receptors in the nervous system [73].
5.2. Environmental Factors
Certain genetic tendencies related to the uptake of folate and B12 increase the risk of autism substantially, but they do not act alone. It has been found that three genetic risk factors for autism: variants in catechol-O-methyl transferase (COMT), methyl tetrahydrofolate reductase (MTHFR), and cystathionine beta synthase (CBS) act in tandem with vitamin depletion to increase autism risk [74]. This is an especially important issue for the many Americans who do not meet the RDA of these vitamins.
Increased use of sunscreen and decreased exposure to the sun may also play a role in the development of autism. One possible mechanism is through vitamin D, which appears to regulate the sodium-sulfate cotransporter NaSi-1. Mice lacking the vitamin D receptor (VDR) had NaSi-1 expression in the kidney which was reduced by 72%, causing urinary sulfate excretion to increase by 42% and serum sulfate concentration to decrease by 50%. In this experiment, levels of hepatic glutathione were reduced by 18% and skeletal sulfated proteoglycans were reduced by 45% [75]. This finding is especially important given the crucial role of elevated NaSi-1 expression in the mother for supplying sufficient amounts of sulfate to the fetus [28]. These findings are consistent with established findings of increased urinary sulfate excretion, decreased serum sulfate, and decreased hepatic glutathione concentrations in autism [7].
In [29] and [76], we proposed that sunlight exposure to the skin is essential not only for vitamin D3 synthesis, but also for the synthesis of sulfate in the skin, and subsequently its combination with cholesterol to form cholesterol sulfate. The skin synthesizes an abundance of cholesterol sulfate, and it has been proposed that the skin is the major supplier of this nutrient to the tissues [77]. In fact, many sunscreens contain a form of retinoic acid (vitamin A) which is a known inhibitor of cholesterol sulfate synthesis by squamous cells in the epidermis [78]. Cholesterol sulfate supply would likely be severely impaired in SLOS–over half of SLOS children are diagnosed with autism, and it is likely that impaired synthesis of cholesterol sulfate in the skin leads to impaired sulfate supply throughout the body. We propose that a similar impairment in the supply of cholesterol sulfate due to the impaired sunlight-stimulated synthesis of sulfate in the skin may be implicated in many other cases of autism.
In fact, there are several epidemiological clues that sunlight exposure in the mother, proposed here to protect against sulfate deficiency, is preventative of autism in the child. Pregnancies which occur in northern latitudes and births timed so that the third trimester corresponds with the late winter or early spring months are more likely to produce a child with autism [79]. In Nordic countries, mothers from foreign backgrounds, who often have darker skin, wear more conservative clothing, and consume reduced amounts of protective foods such as meat and fish, are more likely to have children with autism [80]. For example, Somalian children in Sweden have been found to have rates of autism three to four times higher than in the non-Somali group [81], and this is also thought to be the case for the Somalian population in Minnesota.
Another increasingly common condition associated with a vitamin D deficiency [82] is diabetes, which has recently been shown to be the most important prenatal maternal risk factor for the development of autism [62]. Interestingly, vitamin D3 in the mother is also a risk factor for the development of preeclampsia [83], which, in turn, is also a strong risk factor for future autism in the fetus [84].
In those individuals already compromised by a reduced sulfation capacity, postnatal administration of acetaminophen (branded as androsten, paracetamol or tylenol) could be partially responsible for the rise in autism. Acetaminophen is a xenobiotic which is neutralized and metabolized by the body largely through sulfation, and administration of acetaminophen to rats has been shown to dramatically reduce sulfation capacity to the point of saturation [85]. In fact, acetaminophen use following the measles-mumps-rubella (MMR) vaccine in children has been shown to increase the subsequent risk for autism, and particularly for regressive autism [86]. In the case of children whose sulfation capacities are already stressed and whose central nervous systems are still in the process of developing, it is conceivable that administration of acetaminophen could push the system over the edge and lead to increased damage to the nervous system.
Methionine synthase (MS) activity has been shown to be significantly susceptible to deactivation by numerous environmental toxins [87]. Both heavy metals and the ethylmercury-containing vaccine preservative thimerosol potently interfere with MS activation and impair folate-dependent methylation. Further, it has been demonstrated clinically that elevated mercury body-burden in subjects diagnosed with autism was associated with transsulfuration abnormalities, likely arising from increased oxidative stress and decreased detoxification capacity [88].
6. Reduced Sulfation Capacity Could Explain Other Symptoms Commonly Associated with Autism
Sulfate and the various sulfotransferases play a major role throughout the body, and a deficiency in either has the potential to manifest itself in many ways beyond the neurological defects already described. Indeed, those with autism tend to exhibit peripheral symptoms which may also be associated with a reduced sulfation capacity, including gastrointestinal problems, increased androgen levels, and inflammation. Indeed, these symptoms seem to occur in clusters, and abnormalities in folate-dependent methionine and glutathione metabolism underlying a sulfate deficit have been associated with gastrointestinal and immunologic dysfunction as well as previously discussed damage to the central nervous system [89,90,91].
6.1. Gastrointestinal Problems
It is well known that autism is associated with various gastrointestinal symptoms [92] which have led many to speculate about the role of food sensitivities, dysbiotic intestinal flora, and the gut-brain axis in the pathology of autism [93]. The root of these digestive problems, however, may in some cases be reduced sulfation of the mucous proteins in the gastrointestinal tract which regulate GI tract function [52]. Indeed, sulfated GAGS were found to be decreased in epithelium and basement membrane in the colon of children diagnosed with autism [94]. Reduced sulfation of the proteins has been found to cause inflammation, gut dysfunction, and an increase in gut permeability [95,7], such as is often observed in association with autism spectrum disorders.
6.2. Increased Androgen Levels
The vast majority of the body’s dihydroepiandrosterone (DHEA) is stored as DHEA sulfate, the inactive form, but in the case of decreased sulfation seen in autism, less of the DHEA is converted into DHEA sulfate [51,96], and more is instead converted to testosterone through androstenedione and/or androstenediol. Two recent studies [97,98] reveal elevated androgen levels in subjects diagnosed with autism, as well as substantiating the role of impaired sulfation in inducing the observed elevated levels. Figure 2 illustrates the various sulfation pathways related to important metabolites, where the metabolites that have been identified as present in excess in autism are highlighted in orange [51]. This provides a compelling explanation for the increased testosterone levels seen in both males and females affected with ASD. In turn, the increased levels of testosterone seem to contribute to the pathology of ASD. Testosterone has been found to decrease cystathione β-synthase (CBS) activity, decrease GSH concentrations, and increase susceptibility to oxidative stress [99].
6.3. Inflammation
Depleted cysteine stores, as in the case of autism, stress both sulfate and glutathione synthesis, since both processes compete for cysteine. As expected, this leads to low levels of glutathione (GSH) in autism. Low GSH results in an increase in inflammation, microglial activation, neuroinflammation, and expression of nitric oxide (NO) through inducible nitric oxide synthase (iNOS), all pathologies found in autism [100,101,102].
7. Increasing Sulfate Levels Can Help Alleviate Autistic Symptoms
Although much of the pathology associated with autism seems, unfortunately, to be related to brain development during the critical period and is irreversible, an increase in sulfate levels has, in fact, been shown to alleviate symptoms in some children diagnosed with autism. In one study, a reduction in urinary sulfate excretion achieved through supplementation with molybdenum, an essential metal catalyst in sulphite oxidase, correlated with improvement in autistic symptoms in patients [7]. Oral and transdermal glutathione supplementation, administered to children on the ASD spectrum, led to significant increases in plasma reduced glutathione, sulfate, cysteine, and taurine [103].
In the case of autism, which is hard to pin to one definite cause, it is becoming clearer that many different pathologies lead to the sulfate deficiency which in turn contributes to neurological damage. Depending on the root cause of the sulfate deficiency, different dietary supplements may be beneficial in treating autism. These include, specifically, the vitamins and minerals involved in the transsulfuration pathway (Figure 1): B12, B6, folate, magnesium and zinc. While several of these nutrients have been proposed for the alleviation of autism, findings have been generally inconsistent [104,105]. It is likely that the dysfunctional intestinal epithelium impairs uptake of these nutrients [106].
8. Epigenetics
Epigenetics refers to alterations to the DNA such as methylation at CpG sites or histone acetylation, or the influences of silencing RNAs (siRNAs), which can either increase or decrease expression of the targeted gene [107]. Recent research has led to the realization that epigenetics can greatly influence phenotypic expression, and that the non-coding regions of the genome play an extremely important role in epigenetics. A key epigenetic effect that likely plays a significant if not dominant role in autism is global hypomethylation in the brain, which, we argue, results from impaired methyltransferase/ transsulfuration pathways both in the mother during gestation and in the infant postnatally.
DNA methylation is the first layer of epigenetic effects, and environmental toxins negatively impact DNA methylation globally [11]. Maternal supplementation of folate, particularly in the month before gestation and the first two months of pregnancy, has been demonstrated to have a strong protective effect against autism in the fetus. This could be directly attributed to the important role that folate plays in the methylation pathway. However, one has to be cognizant of the effect that folate will have in redirecting methionine towards methylation pathways rather than towards sulfation pathways, thereby introducing further strain on the sulfation system under conditions of sulfur deprivation.
Experiments on DNA hypomethylation in mouse forebrains have greatly improved our knowledge regarding the effect of global hypomethylation on the developing brain [108]. The experiments involved specifically inducing hypomethylation in excitatory neurons and astrocytes in the dorsal cortex and hippocampus during embryonic development. The modified mice were viable, but they suffered from severe degeneration of the affected brain areas. Microarray analyses revealed that 1047 genes (6.1%) were upregulated, and 444 genes (2.6%) were downregulated. Intriguingly, the mice, while they lived a normal life span, exhibited severe deficits in learning and memory, associated with physical defects such as dystrophic neurites and an increase in dendritic branching.
Several features of these mice correlate with known pathologies associated with autism. Pertubations in sodium and potassium currents were associated with impaired N-methyl-D-aspartate (NMDA) dependent long-term potentiation (LTP), which is also known to be abnormal in association with autism [109]. Sulfate deficiency likely plays a role here as well, since the glycoprotein, N-syndecan, enables LTP by transmitting signals received from factors binding to the attached heparan sulfate proteoglycans [110]. Defective syndecan-3 in mouse hippocampus leads to enhanced LTP and impaired memory [111]. Furthermore, DNA hypomethylation in neural stem cells triggers precocious glial differentiation and activates expression of the glial cell marker, glial fibrillary acidic protein (Gfap) [112], which has been found to be overexpressed in association with autism [113]. This results in an overabundance of glial cells relative to neurons.
Histone deacetylases (HDACs) are enzymes that remove acetyl groups from histones, resulting in the inactivation of the associated genomic region. Valproic acid is an HDAC inhibitor [114] which is used to treat migraine headaches, cancer and mood disorders. Fetal exposure to valproic acid is associated with a highly increased risk to autism [109], suggesting a role for this aspect of epigenetics as well in autism.
Rett syndrome is an X-linked disorder affecting females, which exhibits a behavioral manifestation characteristic of autism [115]. It is caused by a defect in the gene encoding methyl-CpG binding protein 2 (MECP2), which binds to methylated regions and activates the deacetylation of histones, inducing DNA inactivation. MECP2 levels are decreased in the cortex of ASD patients. Thus, impaired MECP2 function resembles impaired methylation in that methylated regions are not properly acted upon.
9. Discussion
Epidemiologic data offer strong support for the hypothesis that prenatal nutrition influences adult susceptibility to disease, and that this phenomenon is likely due to epigenetics [116]. The best known example of this involves the Dutch Hunger Winter of 1944-1945, when war-associated food scarcity led to widespread famine. Children born during this period were more prone to diabetes much later in life [117], and this is likely due to long-lasting epigenetic effects that are initiated during gestation. The budding field of environmental epigenomics addresses the influence that fetal nutritional status and exposure to toxins may have on later risk to conditions like diabetes, obesity and autism [118].
While the milestone of achieving a complete transcription of the genome a decade ago was heralded with great fanfare [119], it was sobering to recognize that the protein coding sequences, the only part we understood, represented a mere 2% of the genome. In the last decade, significant research dollars have been directed towards the goal of decoding the “epigenome,” i.e., to try to understand the crucial role played by the regulatory processes that dictate which genes are activated where and when [120]. The repeated discovery of mutations “in the junk” that influence disease risk [121] forced us to focus on changes in gene activation and suppression in addition to alterations in protein structure due to genetic mutations in coding regions. The topic of most concern to us here is the modifications to gene expression that occur early in development, under the influence of varied nutritional and xenobiotic exposure, which, we argue, can lead to a phenotypical expression that survives throughout life and even into subsequent generations [122].
One of the first identified and most studied epigenetic models is the agouti mouse, where epigenetic effects are readily apparent in their influence on coat color [123]. Agouti mice are born with a range of coat colors, from yellow to dark brown, where yellow is associated with hypomethylation in the promoter region of this retrotransposon during early development, and dark brown with hypermethylation. The yellow coloring is also associated with increased risk to obesity, diabetes, and tumorigenesis [124].
We hypothesize that the agouti gene indirectly encodes the degree of sulfate bioavailability, where the hypomethylated state predicts sulfate deficiency. This follows naturally from the fact that methionine sits at the crossroads of the methylation and transsulfuration pathways. We further hypothesize, as previously proposed [29,76], that sulfate synthesis in the skin is catalyzed by sunlight. The mouse coat color variation is reminiscent of the pale-skinned, blond, blue-eyed Nordic phenotype contrasted with the dark-skinned equatorial African phenotype. Indeed, the agouti gene has been shown to influence the darker skin color of African Americans [125].
We have previously proposed that endothelial nitric oxide synthase (eNOS) is a moonlighting enzyme whose main product is sulfate, catalyzed by sun exposure, and, further, that the sulfate is combined with cholesterol to produce cholesterol sulfate in the skin, which is then transported to all the tissues. The enzyme’s physical structure suggests that cobalamin may facilitate the reaction, as it fits snugly into the heme pocket of NOS isoforms, and is known to inhibit nitric oxide synthesis [126]. eNOS switches to nitric oxide (and hence nitrate) synthesis following calcium entry, calmodulin binding and phosphorylation [127], which we view as a pathological response. eNOS is membrane-bound in caveolae in its so-called “inactive” state, and caveolin suppresses nitric oxide synthesis.
Nitrate can substitute for sulfate for ionic buffering in the blood, but the resulting global sulfate deficiency has pathological consequences [76]. Indeed, autism is associated with excess nitrite and nitrate in the blood [128,129]. We hypothesize that the 1,047 genes that were upregulated and the 444 genes that were downregulated as a consequence of hypomethylation during embryogenesis [108] have to do with adjusting system-level parameters to account for severe sulfate deficiencies. One of those parameters is captured in the agouti gene, which produces a prototype more sensitive to sun exposure in order to increase the sunlight-catalyzed synthesis of sulfate in the skin.
Specific experiments involving the agouti mice are compelling and supportive of our hypothesis. In [123], it was demonstrated that prenatal exposure to the estrogenic environmental toxin, bisphenol A (BPA), induced non-specific gene hypomethylation, leading to favored expression of the yellow phenotype. However, concurrent supplementation with folate, cobalamin, betaine and choline, important nutrients for the supply of methyl groups, abolished this effect. Interestingly, concurrent supplementation with the phytoestrogen, genistein, also abolished the effect. Genistein, a natural estrogen found in soy products, induces mRNA synthesis of both phenol sulfotransferase (hSULT1A1) and human dehydroepiandrosterone (DHEA) sulfotransferase (hSULT2A1) in the liver and colon [130]. DHEA sulfate (DHEAS) is found in relatively high concentrations in the blood stream and can cross the blood-brain barrier [131]. Furthermore, it has been shown that DHEAS, but not DHEA, inhibits GABAA receptors in the nervous system [132]. In fact, other sulfated steroids inhibit GABAA receptors as well in a non-specific way through a mechanism that depends on their generic physical properties as hydrophobic anions [133]. This aligns well with the observed inhibition of these receptors in association with autism [134].
SULT1A1 catalyzes the synthesis of estrone sulfate from estrone, and so would likely enable genistein, a natural estrogen, to transport sulfate from the gut to the nervous system. Thus, both DHEA sulfate and estrone sulfate could substitute for cholesterol sulfate in the blood stream, providing an alternative program for supplying sulfate to the nervous system. We hypothesize that dietary genistein provides sulfate from the digestive system, alleviating the burden on the skin to provide sulfate. BPA, on the other hand, induces DHEA/estrogen synthesis without supplying sulfate, which further burdens the sulfate system due to the need for internally-provided sulfate to transport internally synthesized estrogen.
While folate deficiency is recognized as a risk factor in autism, folate supplementation has met with varied success [135]. Interestingly, these authors propose that there may be a specific relationship between regressive autism and poor response to folate therapy. Furthermore, others have found an autoimmune reaction to folate in the central nervous system in association with autism, which prevents its transport across the blood brain barrier, even when levels are sufficient in the blood stream, associated with the so-called cerebral folate deficiency (CFD) syndrome [136]. In a study of 93 children with autism, over 75% were found to have CFD syndrome, and 44% of the parents who were tested also had the condition.
We hypothesize that all of these observations can be explained if we envision a scenario where the mother is supplemented with folate during gestation, but is severely deficient in supplying cholesterol sulfate to the fetus. The folate will redirect methionine towards methylation pathways rather than sulfotransferase pathways, thus further depleting the already dangerously low sulfate levels. However, the bioavailability of methyl groups will result in a false sense of security, whereby hypermethylated DNA sets up a phenotype that anticipates sulfate sufficiency. It is only after birth and continued subjection to factors that further deplete sulfate, such as lack of sun exposure and toxic environmental chemicals, that the autoimmune reaction to folate develops to spare methionine for its urgent need as a source of sulfate in the brain.
This paper has perhaps produced more questions than answers, but, if our ideas are valid, they provide a framework for interpreting future discoveries of specific genes that are activated/inactivated upon hypomethylation in the brain. These are exciting times, as we are now poised to make significant advances in the field of epigenetics, now that a more detailed accounting of the specific effects of hypomethylation in different tissue types and under differing stimulating conditions is emerging [9]. If we can be allowed to speculate even further, it may well turn out that the genes that have been found to be mutated in association with autism are consequences rather than causes―that these genes are rendered vulnerable because of the underlying nutritional defects, and that this vulnerability is instantiated through epigenetics. We thus hypothesize that epigenetics is in control of evolution, and that hypomethylation causes certain genes to be expressed, and therefore compromised, under the very conditions that promote genetic mutations, such as enhanced oxidation exposure. These are then the genes that are more likely to experience genetic mutations under the adverse environmental conditions that lead to autism. Some of the random mutations may turn out to be beneficial in the context of a world without sunlight.
If our hypothesis is correct, then there is some optimism that reversing the epidemic would not be difficult. If people can be made aware of the importance of sun exposure to the skin to health, then they can alter their lifestyle habits to spend additional time outdoors without sunscreen. Dietary changes involving increases in foods that are good sources of sulfur, such as animal-based proteins and cruciferous vegetables, will help to assure an adequate supply of sulfur, the substrate for sulfate synthesis. Foods that are rich in choline, betaine, folate, cobalamin and zinc would also be encouraged, as these nutrients play an important role in the methylation and sulfotransferase pathways. People should also be encouraged to consume cholesterol-rich foods, as this lessens the burden for cholesterol synthesis, and will likely increase the bioavailability of cholesterol sulfate. Finally, people should make a conscious effort to minimize exposure to xenobioics, which deplete sulfate supplies. Whether the damage can be repaired in individuals already on the autism spectrum is doubtful, but enrichment along these lines would surely be of benefit to them as well.
10. Conclusion
In this paper, we have presented the novel hypothesis that there is a strong link between DNA hypomethylation and sulfate deficiency, both of which have been proposed as key factors associated with autism spectrum disorder. Methionine sits as the crossroads of the methylation and trans-sulfuration pathways, and thus a deficiency in methionine supply impacts both systems. We hypothesize that insufficient sun exposure to the skin compromises the supply of cholesterol sulfate both to the fetus and to the infant postnatally, and that the concurrent exposure to environmental toxins further depletes sulfate and sulfur metabolites. We further hypothesize that hypomethylation during gestation implements a switch towards the production of nitrate rather than sulfate for ionic buffering in the blood stream, and that this has huge negative consequences due to the critical need for sulfate in neuronal maturation. Confirmation of the hypothesis awaits further studies. If our hypothesis is correct, then increased dietary sulfur and increased sun exposure for both the mother and the infant should be strongly encouraged, along with avoidance of exposure to sulfate-depleting environmental toxins.
Acknowledgments
This research was funded in part by Quanta Computers, Taipei, Taiwan, under the auspices of the Qmulus Project, and in part by a grant from the Simons Foundation to the Simons Center for the Social Brain at MIT. The authors are grateful to two reviewers whose efforts resulted in important augmentations from the research literature related to the topic of impaired sulfur metabolism in autism.
References
- Baio, J. Prevalence of Autism Spectrum Disorders Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008; Morbidity and Mortality Weekly Report; Centers for Disease Control and Prevention: Atlanta, GA, 2012.
- Betancur, C.; Leboyer, M.; Gillberg, C. Increased rate of twins among affected sibling pairs with autism. Am. J. Hum. Genet. 2002, 70, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Ferrari, P.; Haimart, M.; Bursztejn, C.; Tabuteau, F.; Braconnier, A.; Pasques-Bondoux, D.; Luong, C. Serotonin Metabolism and other biochemical parameters in infantile autism: A controlled study of 22 autistic children. Neuropsychobiology 1988, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laabs, T.; Carulli, D.; Geller, H.M.; Fawcett, J.W. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr. Opin. Neurobiol. 2005, 15, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Al-Yafee, Y.A.; Al-Ayadhi, L.Y.; Haq, S.H.; El-Ansary, A.K. Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol. 2011, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Grannemann, B.D.; Trivedi, M.H.; Waring, R.H.; Ramsden, D.B.; Garver, C.R. Abnormal sulfation chemistry in autism. In Trends in Autism Research; Ryaskin, O.T., Ed.; Nova Publishers: Hauppauge, NY, USA, 2004; Chapter XI. [Google Scholar]
- Waring, R.H.; Kovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Geiman, T.M.; Muegge, K. DNA methylation in early development. Mol. Reprod. Dev. 2010, 77, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.I.; Lott, P.; Korf, I.; LaSalle, J.M. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011, 21, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Deth, R.; Muratore, C.; Benzecry, J.; Power-Charnitsky, V.-A.; Waly, M. How environmental and genetic factors combine to cause autism: A redox/ methylation hypothesis. Neurotoxicology 2008, 29, 190–201. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2001, 6, 862–869. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla-Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [PubMed]
- Brosnan, J.T.; Jacobs, R.L.; Stead, L.M.; Brosnan, M.E. Methylation demand: A key determinant of homocysteine metabolism. Acta Biochim. Pol. 2004, 51, 405–413. [Google Scholar] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Hubanks, A.; Rose, S.; Gaylor, D.W. Abnormal transmethylation/transsulfuration metabolism and DNA hypomethylation among parents of children with autism. J. Autism Dev. Disord. 2008, 38, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Tsitsiou, E.; Sibley, C.P.; D’Souza, S.W.; Catanescu, O.; Jacobsen, D.W.; Glazier, J.D. Homocysteine is transported by the microvillous plasma membrane of human placenta. J. Inherit. Metab. Dis. 2011, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S- adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J.; et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl deficient cystathionine b-synthase heterozygous mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hines, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and throughout pregnancy predicts fetal homocysteine and birth weight. Clin. Chem. 2000, 50, 1406–1412. [Google Scholar]
- Klaassen, C.D.; Boles, J.W. Sulfation and sulfotransferases 5: The importance of 3’-phosphoadenosine 5’-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997, 11, 404–418. [Google Scholar] [PubMed]
- Salman, E.D.; Kadlubar, S.A.; Falany, C.N. Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain. Drug Metab. Dispos. 2009, 37, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Eagle, K. Toxicological effects of red wine, orange juice, and other dietary SULT1A inhibitors via excess catecholamines. Food Chem. Toxicol. 2012, 50, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Dooley, T.; Obermoeller, R.; Leiter, E. Mapping of the phenol sulfotransferase gene (STP) to human chromosome 16p12.1-p11.2 and to mouse chromosome 7. Genomics 1993, 18, 440–443. [Google Scholar] [PubMed]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E.H., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Shen, Y.; Korn, J. Association between microdeletion and microduplication at 16p11.2 and autism. New Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, P.; Shattock, P. Biochemical aspects in autism spectrum disorders: updating the opioid-excess theory and presenting new opportunities for biomedical intervention. Expert Opin. Ther. Tar. 2002, 6, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Hoffmaster, K.A.; Nezasa, K.; Tallman, M.N.; Brouwer, K.L. Integration of hepatic drug transporters and phase II metabolizing enzymes: Mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur. J. Pharm. Sci. 2006, 27, 447–486. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P. Sulfate in fetal development. Semin. Cell Dev. Biol. 2011, 22, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Mascitelli, L.; Davidson, R. Might cholesterol sulfate deficiency contribute to the development of autistic spectrum disorder? Med. Hypotheses 2012, 8, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Kubushiro, K.; Akiba, Y.; Cui, Y.; Tsukazaki, K.; Nozawa, S.; Iwamori, M. Alteration of acidic lipids in human sera during the course of pregnancy: Characteristic increase in the concentration of cholesterol sulfate. J. Chromatogr. B. Biomed. Sci. Appl. 1997, 704, 99–104. [Google Scholar] [CrossRef]
- Lee, A.; Beck, L.; Brown, R. Identification of a mammalian brain sulfate transporter. Biochem. Biophys. Res. Commun. 1999, 263, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fitzgerald, C.M.; Guptill, J.T.; Booker, A.B.; Kemper, T.L.; Bauman, M.L. Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study. J. Autism Dev. Disord. 2001, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [PubMed]
- Martin, P.T. Glycobiology of the synapse. Glycobiology 2002, 12, 1R–7R. [Google Scholar] [CrossRef] [PubMed]
- Van Vactor, D.; Wall, D.P.; Johnson, K.G. Heparan sulfate proteoglycans and the emergence of neuronal connectivity. Curr. Opin. Neurobiol. 2006, 16, 40–51. [Google Scholar]
- Yamaguchi, Y. Heparan sulfate proteoglycans in the nervous system: Their diverse roles in neurogenesis, axon guidance, and synaptogenesis. Semin. Cell Dev. Biol. 2001, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Bau, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of sulfated glycosaminoglycans in the animal kingdom: Widespread occurrence of heparin-like compounds in invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–289. [Google Scholar] [CrossRef]
- Irie, F.; Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; Vitters, E.L.; de Vries, B J.; van den Berg, W.B. High susceptibility of human articular cartilage glycosaminoglycan synthesis to changes in inorganic sulfate availability. J. Orthoped. Res. 1990, 8, 565–571. [Google Scholar]
- Leboyer, M.; Philippe, A.; Bouvard, M.; Guilloud-Bataille, M.; Bondoux, D.; Tabuteau, F.; Feingold, J.; Mouren-Simeoni, M.C.; Launay, J.M. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiat. 1999, 45, 158–163. [Google Scholar] [CrossRef]
- Minderaa, R.B.; Anderson, G.M.; Volkmar, F.R.; Akkerhuis, G.W.; Cohen, D.J. Urinary 5-hydroxyindoleacetic acid and whole blood serotonin and tryptophan in autistic and normal subjects. Biol. Psychiat. 1987, 22, 933–940. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. Serotonin and brain development: Role in human developmental diseases. Brain Res. Bull. 2001, 56, 479–485. [Google Scholar] [CrossRef]
- Whitaker-Azmitia, P.M. Behavioral and cellular consequences of increasing serotonergic activity during brain development: A role in autism? Int. J. Dev. Neurosci. 2005, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B. Neuroinflammation, blood-brain barrier, seizures and autism. J. Neuroinflam. 2011, 8, 168. [Google Scholar]
- Green, L.; Fein, D.; Modahl, C.; Feinstein, C.; Waterhouse, L.; Morris, M. Oxytocin and autistic disorder: alterations in peptide forms. Biol. Psychiat. 2001, 50, 609–613. [Google Scholar] [CrossRef]
- Davis, E.; Fennoy, I.; Laraque, D.; Kanem, N.; Brown, G.; Mitchell, J. Autism and developmental abnormalities in children with perinatal cocaine exposure. J. Natl. Med. Assoc. 1988, 84, 315–319. [Google Scholar]
- Aneja, A.; Tierney, E. Cholesterol deficit in autism: Insights from Smith-Lemli-Opitz syndrome. 2008. [Google Scholar] [CrossRef]
- Scanlon, S.M.; Williams, D.C.; Schloss, P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry 2001, 40, 10507–10513. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Geier, M.R. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem. Res. 2009, 34, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P.; Nemes, B.; Vlase, L.; Gagyi, C.E.; Dronca, E.; Miu, A.C.; Dronca, M. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006, 78, 2244–2448. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Geier, M.R. A clinical and laboratory evaluation of methionine cycle- transsulfuration and androgen pathway markers in children with autistic disorders. Horm. Res. Pediatr. 2006, 66, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Pirrone, P.; Elia, M.; Waring, R.H.; Romano, C. Sulphation deficit in ‘low-functioning’ autistic children: a pilot study. Biolog. Psychiat. 1999, 46, 420–424. [Google Scholar] [CrossRef]
- O’Reilly, B.A.; Waring, R.H. Enzyme and sulphur oxidation deficiencies in autistic children with known food/chemical intolerances. J. Orthomol. Med. 1993, 8, 198–200. [Google Scholar]
- Lee, A.; Dawson, P.A.; Markovich, D. NaSi-1 and Sat-1: Structure, function and transcriptional regulation of two genes encoding renal proximal tubular sulfate transporters. Int. J. Biochem. Cell. Biol. 2005, 37, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Melnyk, S.; Jernigan, S. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B 2006, 141B, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Azeim, S.; Li, X.; Chung, L.W.; Morokuma, K. Zinc-homocysteine binding in cobalamin-dependent methionine synthase and its role in the substrate activation: DFT, ONIOM, and QM/MM molecular dynamics studies. J. Comp. Chem. 2011, 32, 3154–3167. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef] [PubMed]
- Matherly, L.H. Molecular and cellular biology of the human reduced folate carrier. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 131–162. [Google Scholar] [PubMed]
- Yates, Z.; Lucock, M. G80A reduced folate carrier SNP modulates cellular uptake of folate and affords protection against thrombosis via a non homocysteine related mechanism. Life Sci. 2005, 77, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Melnyk, S. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Karatela, R.; Sainani, G. Plasma homocysteine in obese, overweight and normal weight hypertensives and normotensives. Indian Heart J. 2009, 61, 156–159. [Google Scholar] [PubMed]
- Krakowiak, P.; Walker, C.K.; Bremer, A.A.; Baker, A.S.; Ozonoff, S.; Hansen, R.L.; Hertz-Picciotto, I. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012, 129, e1121–e1128. [Google Scholar] [CrossRef] [PubMed]
- Seetharam, B.; Bose, S.; Li, N. Cellular import of cobalamin. J. Nutr. 1999, 129, 1761–1764. [Google Scholar] [PubMed]
- Grattan-Smith, P.J.; Wilcken, B.; Procopis, P.G.; Wise, G.A. The neurological syndrome of infantile cobalamin deficiency: Developmental regression and involuntary movements. Mov. Disord. 1997, 12, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. The role of paraoxonase 1 in the detoxification of homocysteine thiolactone. Adv. Exp. Med. Biol. 2010, 660, 113–127. [Google Scholar] [PubMed]
- McCully, K.S. Chemical pathology of homocysteine V: thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Annals Clin. Lab. Sci. 2011, 41, 300–313. [Google Scholar]
- Dufault, R.; Lukiw, W.J.; Crider, R.; Schnoll, R.; Wallinga, D.; Deth, R. A macroepigenetic approach to identify factors responsible for the autism epidemic in the United States. Clin. Epigenet. 2012, 4, 6. [Google Scholar]
- Ackerman, Z.; Oron-Herman, M.; Pappo, O.; Peleg, E.; Safadi, R.; Schmilovitz-Weiss, H.; Grozovski, M. Hepatic effects of rosiglitazone in rats with the metabolic syndrome. Basic Clin. Pharmacol. Toxicol. 2010, 107, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [PubMed]
- Alarcón, M.; Abrahams, B. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [PubMed]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 409, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Sakurai, T.; Buxbaum, J.D. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Hansen, R.; Hartiala, J. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology 2011, 22, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.J.G.; Liu, W.; Qiao, G.; Kong, J.; Zheng, W.; Krausz, T.; Cs-Szabo, G.; Sitrin, M.D.; Li, Y.C. Critical role of vitamin D in sulfate homeostasis: Regulation of the sodium-sulfate cotransporter by 1,25-dihydroxyvitamin D3. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E744–E749. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.M.; Seneff, S. The initial common pathway of inflammation, disease, and sudden death. Entropy 2012, 14, 1399–1442. [Google Scholar] [CrossRef] [Green Version]
- Strott, C.A. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Rearick, J.I.; Stoner, G.D.; George, M.A.; Jetten, A.M. Cholesterol Sulfate Accumulation in Tumorigenic and Nontumorigenic Rat Esophageal Epithelial Cells: Evidence for Defective Differentiation Control in Tumorigenic Cells. Cancer Res. 1988, 48, 5289–5295. [Google Scholar] [PubMed]
- Grant, W.B.; Soles, C.M. Epidemiologic evidence supporting the role of maternal vitamin D deficiency as a risk factor for the development of infantile autism. Dermatoendocrinol. 2009, 1, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Prenatal risk factors for autism: comprehensive meta-analysis. Brit. J. Pschiat. 2009, 195, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barnevik-Olsson, M.; Gillberg, C.; Fernell, E. Prevalence of autism in children born to Somali parents living in Sweden: A brief report. Dev. Med. Child Neurol. 2008, 50, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Hughes, B. The role of vitamin D and calcium in type 2 diabetes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Catov, J.M.; Simhan, H.N.; Holick, M.F.; Powers, R.W.; Roberts, J.M. Maternal vitamin D deficiency increases the risk of preeclampsia. J. Clin. Endocrinol. Metab. 2007, 92, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; McDermott, S.; Bao, H.; Hardin, J.; Gregg, A. Preeclampsia, birth weight, and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Klaassen, C.D. Different mechanism of saturation of acetaminophen sulfate conjugation in mice and rats. Toxicol. Appl. Pharmacol. 1996, 139, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.T.; Klonoff-Cohen, H.S.; Wingard, D.L.; Akshoomoff, N.A.; Macera, C.A.; Ji, M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: The results of a parent survey. Autism 2008, 12, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Waly, M.; Olteanu, H.; Banerjee, R.; Choi, S.W.; Mason, J.B.; Parker, B.S.; Sukumar, S.; Shim, S.; Sharma, A.; Benzecry, J.M.; et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Mol. Psychiatry 2004, 9, 358–370. [Google Scholar] [PubMed]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Nataf, R.; Geier, M.R. Biomarkers of environmental toxicity and susceptibility in autism. J. Neurol. Sci. 2009, 280, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, R.; Pittack, N.; Nebe, C.T.; Schuster, D.; Brust, J.; Beichert, M.; Hack, V.; Daniel, V.; Edler, L.; Dröge, W. Improvement of immune functions in HIV infection by sulfur supplementation: Two randomized trials. J. Mol. Med. (Berl.). 2000, 78, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Breitkreutz, R. Glutathione and immune function. Proc. Nutr. Soc. 2000, 59, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Jain, A.; Meister, A. Glutathione is required for intestinal function. Proc. Natl. Acad. Sci. USA 1990, 87, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.; Perman, J.A. Autism and gastrointestinal symptoms. Curr. Gastroenterol. Rep. 2002, 4, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Doyle, R.; Francis, K.; Conti, P.; Kalogeromitros, D. Novel therapeutic targets for autism. Trends Pharmacol. Sci. 2008, 29, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Furlano, R.I.; Anthony, A.; Day, R.; Brown, A.; McGarvey, L.; Thomson, M.A.; Davies, S.E.; Berelowitz, M.; Forbes, A.; Wakefield, A.J.; et al. Colonic CD8 and γδ T-cell infiltration with epithelial damage in children with autism. J. Pediatr. 2001, 138, 366–372. [Google Scholar]
- Murch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Levin, M.; Lionetti, P.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar]
- Strous, R.D.; Golubchik, P.; Maayan, R.; Mozes, T.; Tuati-Werner, D.; Weizman, A.; Spivak, B. Lowered DHEA-S plasma levels in adult individuals with autistic disorder. Eur. Neuropsychopharmacol. 2005, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Geier, M.R. A prospective assessment of androgen levels in patients with autistic spectrum disorders: Biochemical underpinnings and suggested therapies. Neuro. Endocrinol. Lett. 2007, 28, 565–573. [Google Scholar] [PubMed]
- Geier, D.A.; Kern, J.K.; King, P.G.; Sykes, L.K.; Geier, M.R. An evaluation of the role and treatment of elevated male hormones in autism spectrum disorders. Acta Neurobiol. Exp. (Wars). 2012, 72, 1–17. [Google Scholar] [PubMed]
- Prudova, A.; Albin, M.; Bauman, Z.; Lin, A.; Vitvitsky, V.; Banerjee, R. Testosterone Regulation of Homocysteine metabolism modulates redox status in human prostate cancer cells. Antiox. Redox Sig. 2007, 9, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P. The immune response in autism: A new frontier for autism research. J. Leukoc. Biol. 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.L.; Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 2009, 16, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Adams, J.B.; Garver, C.R.; Audhya, T.; Geier, M.R. A clinical trial of glutathione supplementation in autism spectrum disorders. Med. Sci. Monit. 2011, 17, CR677–C682. [Google Scholar] [CrossRef] [PubMed]
- Martineau, J.; Barthelemy, C.; Garreau, B.; Lelord, G. Vitamin B6, magnesium, and combined B6-Mg: Therapeutic effects in childhood autism. Biol. Psychiatry 1985, 20, 467–478. [Google Scholar] [CrossRef]
- Mousain-Bosc, M.; Roche, M.; Polge, A.; Pradal-Prat, D.; Rapin, J.; Bali, J.P. Improvement of neurobehavioral disorders in children supplemented with magnesium-vitamin B6. II. Pervasive developmental disorder-autism. Magnesium Res. 2006, 19, 53–62. [Google Scholar]
- Allen, L.H. Causes of vitamin B12 and folate deficiency. Food Nutr. Bull. 2008, 29, 20–34. [Google Scholar]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, L.K.; Golshani, P.; Namihira, M.; Xue, Z.; Matynia, A.; Yang, W.X.; Silva, A.J.; Schweizer, F.E.; Fan, G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 2009, 18, 2875–2888. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed]
- Lauri, S.E.; Kaukinen, S.; Kinnunen, T.; Ylinen, A.; Imai, S.; Kaila, K.; Taira, T.; Rauvala, H. Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J. Neurosci. 1999, 19, 1226–12235. [Google Scholar] [PubMed]
- Kaksonen, M.; Pavlov, I.; Voikar, V.; Lauri, S.E.; Hienola, A.; Riekki, R.; Lakso, M.; Taira, T.; Rauvala, H. Syndecan-3-deficient mice exhibit enhance LTP and impaired hippocampus-dependent memory. Mol. Cell Neurosci. 2002, 21, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Martinowich, K.; Chin, M.H.; He, F.; Fouse, S.D.; Hutnick, L.; Hattori, D.; Ge, W.; Shen, Y.; Wu, H.; et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 2005, 132, 3345–3356. [Google Scholar] [PubMed]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krömer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo-Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–69678. [Google Scholar] [PubMed]
- Woods, R.; Vallero, R.O.; Golub, M.S.; Suarez, J.K.; Ta, T.A.; Yasui, D.H.; Chi, L.H.; Kostyniak, P.J.; Pessah, I.N.; Berman, R.F.; LaSalle, J.M. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum. Mol. Genet. 2012, 21, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 2004, 20, 63–68. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, S.R.; Painter, R.C.; Phillips, D.I.W.; Osmond, C.; Michels, R.P.J.; Godsland, I.F.; Bossuyt, P.M.M.; Bleker, O.P.; Roseboom, T.J. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 2006, 29, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L. Controversial from the start. Science 2001, 291, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Biémont, C.; Vieira, C. Genetics: Junk DNA as an evolutionary force. Nature 2006, 443, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Chong, S.; Champ, M.E.; Cuthbert, P.C.; Morgan, H.D.; Luu, K.V.K.; Whitelaw, E. Transgenerational inheritance of epigenetic states at the murine AxinFu allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 2538–2543. [Google Scholar]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Miltenberger, R.; Mynatt, R.; Wilkinson, J.; Woychik, R. The role of the agouti gene in the yellow obese syndrome. J. Nutr. 1997, 127, S1902–S1907. [Google Scholar]
- Bonilla, C.; Boxill, L.A.; Donald, S.A.; Williams, T.; Sylvester, N.; Parra, E.J.; Dios, S.; Norton, H.L.; Shriver, M.D.; Kittles, R.A. The 8818G allele of the agouti signaling protein (ASIP) gene is ancestral and is associated with darker skin color in African Americans. Hum. Genet. 2005, 116, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Chen, Y.; Jiang, N.; Beasley, B.E.; Salerno, J.C.; Ghosh, D.K. Inhibition of nitric oxide synthase by cobalamins and cobinamides. Free Radic. Biol. Med. 2009, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.-P.; Sessa, W.C. Post-translational control of endothelial nitric oxide synthase: Why isn’t calcium/calmodulin enough? J. Pharmacol. Exp. Ther. 2001, 299, 818–824. [Google Scholar] [PubMed]
- Sögüt, S.S.; Zoroglu, S.S.; Özyurt, H.; Ylmaz, H.R.; Ozugurlu, F.; Sivasli, E.; Yetkin, O.; Yanik, M.; Tutkun, H.; Savas, H.A.; et al. Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin. Chim. Acta 2003, 331, 111–117. [Google Scholar] [PubMed]
- Akyol, O.; Zoroglu, S.S.; Armutcu, F.; Sahin, S.; Gurel, A. Nitric Oxide as a Physiopathological Factor in Neuropsychiatric Disorders. In Vivo 2004, 18, 377–390. [Google Scholar] [PubMed]
- Chen, Y.; Huang, C.; Zhou, T.; Chen, G. Genistein induction of human sulfotransferases in HepG2 and Caco-2 cells. Basic Clin. Pharmacol. Toxicol. 2008, 103, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Asaba, H.; Hosoya, K.; Takanaga, H.; Ohtsuki, S.; Tamura, E.; Takizawa, T.; Terasaki, T. Blood-brain barrier is involved in the efflux transport of a neuroactive steroid, dehydroepiandrosterone sulfate, via organic anion transporting polypeptide 2. J. Neurochem. 2000, 75, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Roh, D.H.; Seo, H.S.; Kang, S.Y.; Han, H.J.; Beitz, A.J.; Lee, J.H. Intrathecal injection of the neurosteroid, DHEAS, produces mechanical allodynia in mice: Involvement of spinal sigma-1 and GABAA receptors. Br. J. Pharmacol. 2009, 157, 666–773. [Google Scholar]
- Chisari, M.; Wu, K.; Zorumski, C.F.; Mennerick, S. Hydrophobic anions potently and uncompetitively antagonize GABA(A) receptor function in the absence of a conventional binding site. Br. J. Pharmacol. 2011, 164, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Menold, M.M.; Shao, Y.; Wolpert, C.M.; Donnelly, S.L.; Raiford, K.L.; Martin, E.R.; Ravan, S.A.; Abramson, R.K.; Wright, H.H.; Delong, G.R.; et al. Association analysis of chromosome 15 GABAA receptor subunit genes in autistic disorder. J. Neurogenet. 2001, 15, 245–259. [Google Scholar] [PubMed]
- Goin-Kochel, R.P.; Porter, A.E.; Peters, S.U.; Shinawi, M.; Sahoo, T.; Beaudet, A.L. The MTHFR 677C-T polymorphism and behaviors in children with autism: Exploratory genotypephenotype correlations. Autism Res. 2009, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol. Psychiatr. 2012. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Hartzell, S.; Seneff, S. Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? Entropy 2012, 14, 1953-1977. https://doi.org/10.3390/e14101953
AMA Style
Hartzell S, Seneff S. Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism? Entropy. 2012; 14(10):1953-1977. https://doi.org/10.3390/e14101953
Chicago/Turabian StyleHartzell, Samantha, and Stephanie Seneff. 2012. "Impaired Sulfate Metabolism and Epigenetics: Is There a Link in Autism?" Entropy 14, no. 10: 1953-1977. https://doi.org/10.3390/e14101953