1. Introduction
Bimolecular nucleophilic substitution (S
N2) reactions at carbon centers continue to be among the most studied chemical reactions. Early research was only restricted to solution phase chemistry, but with the advent of modern experimental techniques [
1,
2] gas-phase chemistry has gained much interest for S
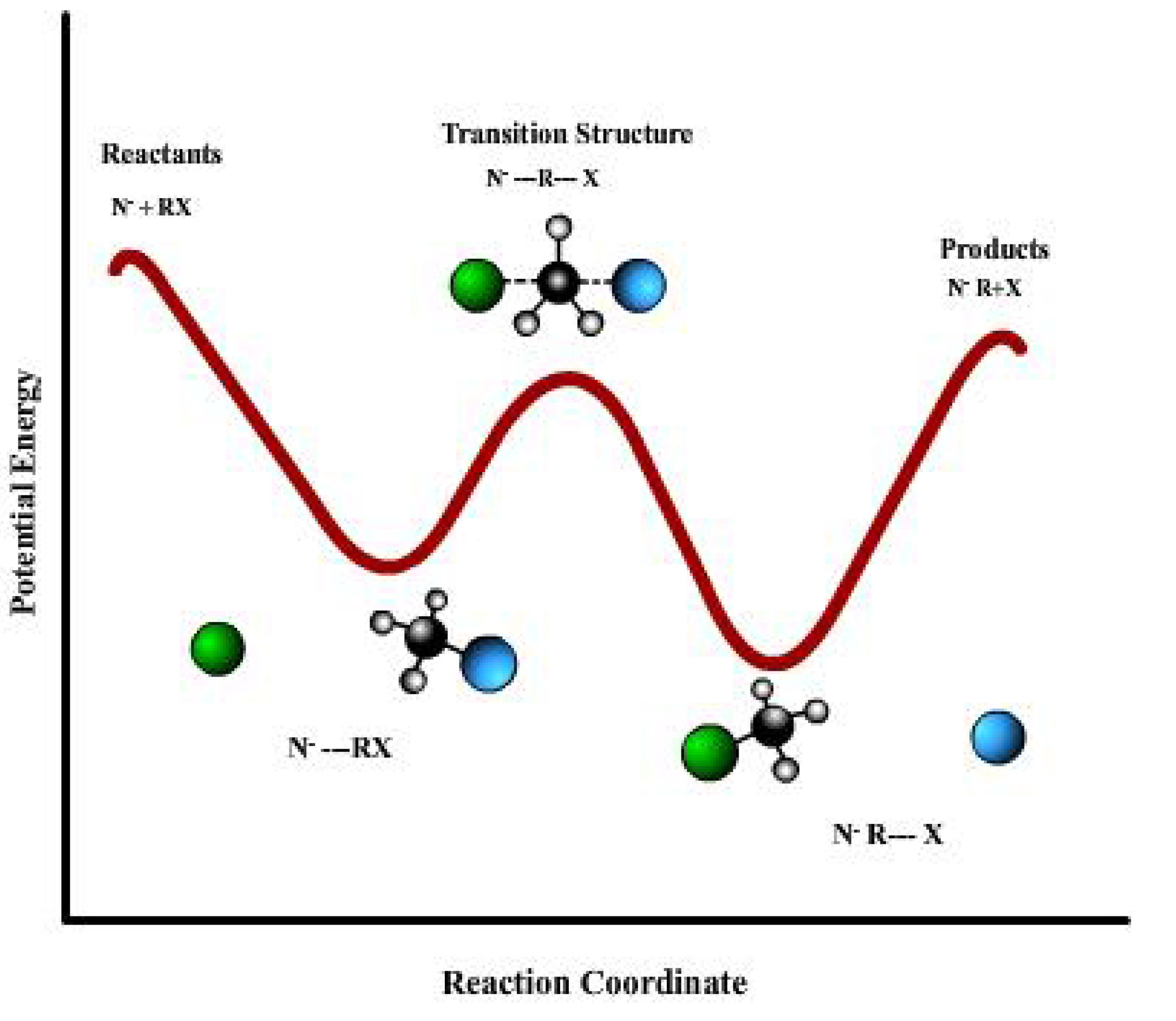
N2 reactions. It was discovered that these reactions generally exhibit a double-well potential with a central barrier, as depicted in
Figure 1 for the reaction of
N− and R
X, where
N− stands for the nucleophile and
X for the nucleofuge. The double-well feature for S
N2 reactions has been the subject of a great deal of research on the theoretical side by use of powerful computers coupled with modern techniques of molecular electronic structure. Recently gas-phase S
N2 reactions have been widely investigated by kinetic experiments [
3,
4,
5], ab initio quantum and semiclassical dynamical methods and trajectory simulations [
6,
7], statistical mechanical studies [
8,
9],
ab-initio and density functional structural analyses [
10,
11,
12,
13,
14]. The importance of S
N2 studies in the chemical literature has made these reactions to constitute a paradigm for quantitative understandings of ion molecule reactions in general [
15].
Figure 1.
The general shape of the potential energy for a SN2 reaction. Reactants and products, at the beginning and the end of the chemical path, are connected by the TS and two minima which indicates the ionic complexes.
Figure 1.
The general shape of the potential energy for a SN2 reaction. Reactants and products, at the beginning and the end of the chemical path, are connected by the TS and two minima which indicates the ionic complexes.
Studying the energetics of chemical reactions has been a prime goal for theoretical chemistry over the last decades [
16]. For instance, a variety of calculations of potential energy surfaces have been performed at various levels of sophistication [
17]. Within these efforts, particular interest has been focused on extracting chemically meaningful information about the stationary points of the potential energy surface (PES). Despite the fact that minima, maxima, and saddle points are useful mathematical features of the energy surface to keep tracking chemical reaction-paths [
18], it has been difficult to attribute too much chemical or physical meaning to these critical points [
19,
20].
In connection to the above, transition-state theory has achieved widespread acceptance as a tool for the interpretation of chemical reaction rates and has led to much insight into chemical and physical processes [
21]. The fundamental assumptions involved in transition state (TS) theory are that molecules must traverse saddle points (transition states) of the potential energy surface. Hence, the TS is the focus of the entire theory. The pursuit for understanding the transition state (TS) represents a challenge of physical organic chemistry. Many efforts to achieve the latter have produced chemically useful descriptions of the TS, such as the one provided by Hammond and Leffler [
22,
23]. Whereas the reaction rate and the reaction barrier are chemical concepts, which have been rigorously defined and experimentally studied since the origin of the TS theory [
24,
25] the comprehension of the TS structure remains to be a challenge for physical organic chemists. Therefore, understanding the TS is a fundamental goal of chemical reactivity theories, which implies the knowledge of the chemical events that take place in its vicinity to better understand the reaction.
On the other hand femtochemistry [
26,
27] has yielded one of the most promising techniques, the anion photodetachment spectra [
28], through which the direct observation of TS has been possible. The femto-time scale is perhaps the ultimate frontier as far as chemistry is concerned, experiments at this level being ultimately limited by the fundamental laws of quantum mechanics through the Heisenberg principle. To explain the experimental results of the femtotechniques, it will be necessary to complement the existing chemical reactivity theories with electronic density descriptors of the events taking place in the vicinity of the transition-state region, where the chemical bonds are actually being formed or destroyed.
In connection with the above, there are a number of studies in the literature, which have used a variety of descriptors to study either the TS or to follow the course of the chemical reaction path. For instance, Shi and Boyd [
29] performed a systematic analysis of model S
N2 reactions to study the TS charge distribution in connection with the Hammond-Leffler postulate. Bader and MacDougall [
30] developed a theory of reactivity based on the Laplacian of the charge density so as to align the local charge concentrations with regions of charge depletion of the reactants, and hence mixing in the lowest energy excited state of the combined system to produce a relaxed charge distribution corresponding to the transition state density. By studying the time evolution of a bimolecular exchange reaction Balakrishnan et al showed that information-theoretic entropies in dual or phase space rose to a maximum in a dynamical study [
31]. In an attempt to build a density-based theory of chemical reactivity, Knoerr and Eberhart [
32] reported correlations between features of the quantum-mechanically determined charge density and the energy-based measures of Shaik and collaborators to describe the charge transfer, stability, and charge localization accompanying an S
N2 reaction [
33]. Moreover, Tachibana [
34] was able to visualize the formation of a chemical bond of selected model reactions by using the kinetic energy density to identify the intrinsic shape of the reactants, the TS and the reaction products along the course of the IRC, hence realizing Coulson’s conjecture [
35] in that the physical meaning of the probability density might be related with the energy density. The reaction force of a system’s potential energy along the reaction coordinate has been used to characterize changes in the structural and/or electronic properties in chemical reactions [
36,
37,
38,
39]. The Kullback–Leibler information deficiency has been evaluated along molecular internal rotational or vibrational coordinates and along the intrinsic reaction coordinate for several S
N2 reactions [
40]. In past years, there has been an increasing interest to analyze the electronic structure of atoms and molecules by applying information theory (IT) [
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64]. In recent studies, we have shown that information-theoretic measures are capable of providing simple pictorial chemical descriptions of atoms and molecules. For instance, information theoretical analyses have shown to be useful for the phenomenological description of the course of elementary chemical reactions. This is achieved through the
localized/delocalized behavior of the electron densities in position and momentum spaces by revealing important chemical regions that are not present in the energy profile, such as the ones in which bond forming and bond breaking occur and also where bond cleavage energy regions (BCER) appear. Furthermore, the synchronous reaction mechanism of a S
N2 type chemical reaction and the non-synchronous behavior of the simplest hydrogen abstraction reaction were predicted by use of Shannon entropies analysis. Recent reviews of information-theoretical analysis of elementary chemical reactins and their concurrent phenomena have been published recently [
65,
66].
Moreover, the application of complexity concepts in physical sciences has acquired increasing interest over the last years. The definition of complexity is not unique, its quantitative characterization has been an important subject of research and it has received considerable attention [
67]. The usefulness of each definition depends on the type of system or process under study, the level of the description, and the scale of the interactions among either elementary particles, atoms, molecules, biological systems,
etc. Fundamental concepts such as uncertainty or randomness are frequently employed in the definitions of complexity, although some other concepts like clustering, order, localization or organization might be also important for characterizing the complexity of systems or processes. It is not clear how the aforementioned concepts might intervene in the definitions so as to quantitatively assess the complexity of the system. However, recent proposals have formulated this quantity as a product of two factors, taking into account
order/disequilibrium and
delocalization/uncertainty. This is the case of the definition of López-Mancini-Calbet (LMC) shape complexity [
68,
69,
70,
71,
72] that, like others, satisfies the boundary conditions by reaching its minimal value in the extreme ordered and disordered limits. The LMC measure is constructed as the product of two important information-theoretic quantities (see below): the so-called disequilibrium
D (also known as self-similarity [
73] or information energy [
74]), which quantifies the departure of the probability density from uniformity [
71,
72] (equiprobability) and the Shannon entropy S, which is a general measure of
delocalization/uncertainty of the probability density [
68], and quantifies the departure of the probability density from localizability. Both global quantities are closely related to the measure of spread of a probability distribution. The Fisher-Shannon product
FS has been employed as a measure of atomic correlation [
51] and also defined as a statistical complexity measure [
75,
76]. The product of the power entropy J-explicitly defined in terms of the Shannon entropy- (see below) and the Fisher information measure,
I, combine both the global character (considering the distribution as a whole) and the local one (in terms of the gradient of the distribution), to preserve the general complexity properties. As compared to the LMC complexity, aside of the explicit dependence on the Shannon entropy which serves to measure the uncertainty (localizability) of the distribution, the Fisher-Shannon complexity replaces the disequilibrium global factor
D by the Fisher local one to quantify the departure of the probability density from disorder [
77,
78] of a given system through the gradient of the distribution.
In the present study we have undertaken an information-complexity analysis of the SN2 exchange reaction for CH3Cl + F− by use of two-component planes as well as the LMC and FS complexity products. The goals of the study are three-fold: (i) to interpret the information features of the double-well potential with a central barrier exhibited by these kind of reactions (SN2 exchange reaction); (ii) to analyze the charge transfer process that is developed along the IRC of the CH3Cl + F− exchange reaction; (iii) to characterize the concurrent phenomena occurring at the vicinity of the TS. Our interest will be focused on the recognition of patterns of uncertainty/localizability, disorder/narrowness and disequilibrium/uniformity through the statistical complexity analysis.
The organization of the paper is as follows: in
Section 2 we defined the complexity measures along with their information-theoretic components. In
Section 3 we calculate the information components as well as the Fisher-Shannon and LMC complexities. These information functionals of the one-particle density are computed in position (r) and momentum (p) spaces. Besides, the Fisher-Shannon (
I-J) and the disequilibrium-Shannon (
D-L) planes are studied. In
Section 4, some conclusions are given.
2. Information-Theoretical Measures and Complexities
In the independent-particle approximation, the total density distribution in a molecule is a sum of contribution from the electrons in each of the occupied orbitals. This is the case in both
r-space and
p-space, position and momentum respectively. In momentum space, the total electron density,
, is obtained through the molecular momentals (momentum-space orbitals)
, and similarly for the position-space density,
, through the molecular position-space orbitals
(Atomic units (a.u.) are used throughout). The momentals can be obtained by three-dimensional Fourier transformation of the corresponding orbitals (and conversely):
Standard procedures for the Fourier transformation of position space orbitals generated by
ab-initio methods have been described [
77]. The orbitals employed in
ab-initio methods are linear combinations of atomic basis functions and since analytic expressions are known for the Fourier transforms of such basis functions [
78], the transformation of the total molecular electronic wave function from position to momentum space is computationally straightforward [
79].
As we mentioned in the introduction, the
LMC-shape complexity is defined through the product of two relevant information-theoretic measures. So that, for a given probability density in position space,
, the C(
LMC) complexity is given by [
67,
69,
70,
71,
72,
73,
74,
80,
8182]:
where
Dr is the disequilibrium [
73,
74]:
and
S is the Shannon entropy [
68]:
from which the exponential entropy
is defined. Similar expressions for the LMC complexity measure in the conjugated momentum space might be defined for a distribution
:
It is important to mention that the
LMC complexity of a system must comply with the following lower bound [
83]:
The FS complexity in position space,
Cr(FS), is defined in terms of the product of the Fisher information [
84,
85]:
and the power entropy [
36,
37] in position space,
Jr:
which depends on the Shannon entropy defined above. So that, the
FS complexity in position space is given by:
and similarly:
in momentum space.
Let us remark that the factors in the power Shannon entropy
J are chosen to preserve the invariance under scaling transformations, as well as the rigorous relationship [
86]:
with
n being the space dimensionality, thus providing a universal lower bound to
FS complexity. The definition in Equation (8) corresponds to the particular case
n = 3, the exponent containing a factor
2/n for arbitrary dimensionality. It is worthwhile noting that the aforementioned inequalities remain valid for distributions normalized to unity, which is the choice that it is employed throughout this work for the 3-dimensional molecular case.
3. Entropy and Complexity Measures
In order to associate the aforementioned information-theoretical measures along the course of the reaction path, we have also employed some selected physical descriptors: the atomic charges obtained from the molecular electrostatic potential (MEP) and the atomic electric potentials associated with the basins of the MEP (see below). Both properties are of physical interest to analyze the frame of coulombic interactions,
i.e., a higher/lower electric potential (being negative) means a lower/higher capacity to acquire negative electron charge. Interestingly, situations of stronger inter-atomic electrostatic interactions or bonding occur when the difference between the atomic electric potentials of two or more species, either atoms or molecular fragments, become equalized [
65].
The MEP represents the molecular potential energy of a proton at a particular location near a molecule [
87], say at nucleus
A. Then, the electrostatic potential,
VA , is defined as:
where
is the molecular electronic distribution and Z
A is the nuclear charge on atom A, located at RA. Generally speaking, negative electrostatic potential corresponds to the attraction of the proton by the concentrated electron density in the molecules from lone pairs, pi-bonds,
etc. Positive electrostatic potential corresponds to repulsion of the proton by the atomic nuclei in regions where low electron density exists and the nuclear charge is incompletely shielded. The most popular methods for extracting charges from molecular wavefunctions are based on fitting of the atomic charges to the MEP computed with ab initio methods. The charge fitting procedure consists of minimizing the root-mean squared deviation between the Coulombic potential generated by the atomic charges and the MEP. These non-bonded atomic electric potentials (ε
i) along with the fitted atomic charges (q
i) will be employed throughout the study by use of the CHELPG method [
88].
The electronic structure calculations performed in the present study were carried out with the Gaussian 09 suite of programs [
89]. Calculations for the IRC were performed at the RHF level of theory with at least 64 points evenly distributed along the reaction path. A high level of theory and a well-balanced basis set (diffuse and polarized orbitals) were then chosen to determine all of the properties for the chemical structures corresponding position to the IRC. Thus, the CISD method was employed in addition to the 6-311++G(2d,3p) basis set, unless otherwise stated. All information theoretical quantities are determined in position space except for the
FS and the
LMC-shape complexities which were calculated in both spaces, for all the 64 chemical structures along the IRC of the reaction by use of a suite of programs developed in our laboratory along with 3D numerical integration routines [
90,
91], and the DGRID software package [
79].
In recent research we have studied chemical reactivity under the information-theoretic perspective [
65,
66]. It is worth noting that information theory has proved to be useful for the phenomenological description of concurrent processes in elementary chemical reactions;
i.e., the bond breaking/forming region (
B-B/F), bond cleavage energy region (BCER), transition state (TS) among others, are revealed under information- theoretic measures. It is worth mentioning that the energy profile of elementary reactions does not describe all chemically significant features of the course of these reactions. The latter is perhaps the essential argument which justifies the entire analysis on the information-theoretical side, taking into account the apparent success that this approach has achieved for the chemical description of reactivity phenomena [
65,
66]. The energy profile for a typical S
N2 substitution reaction is depicted in
Figure 1.
The reaction can be represented as:
In
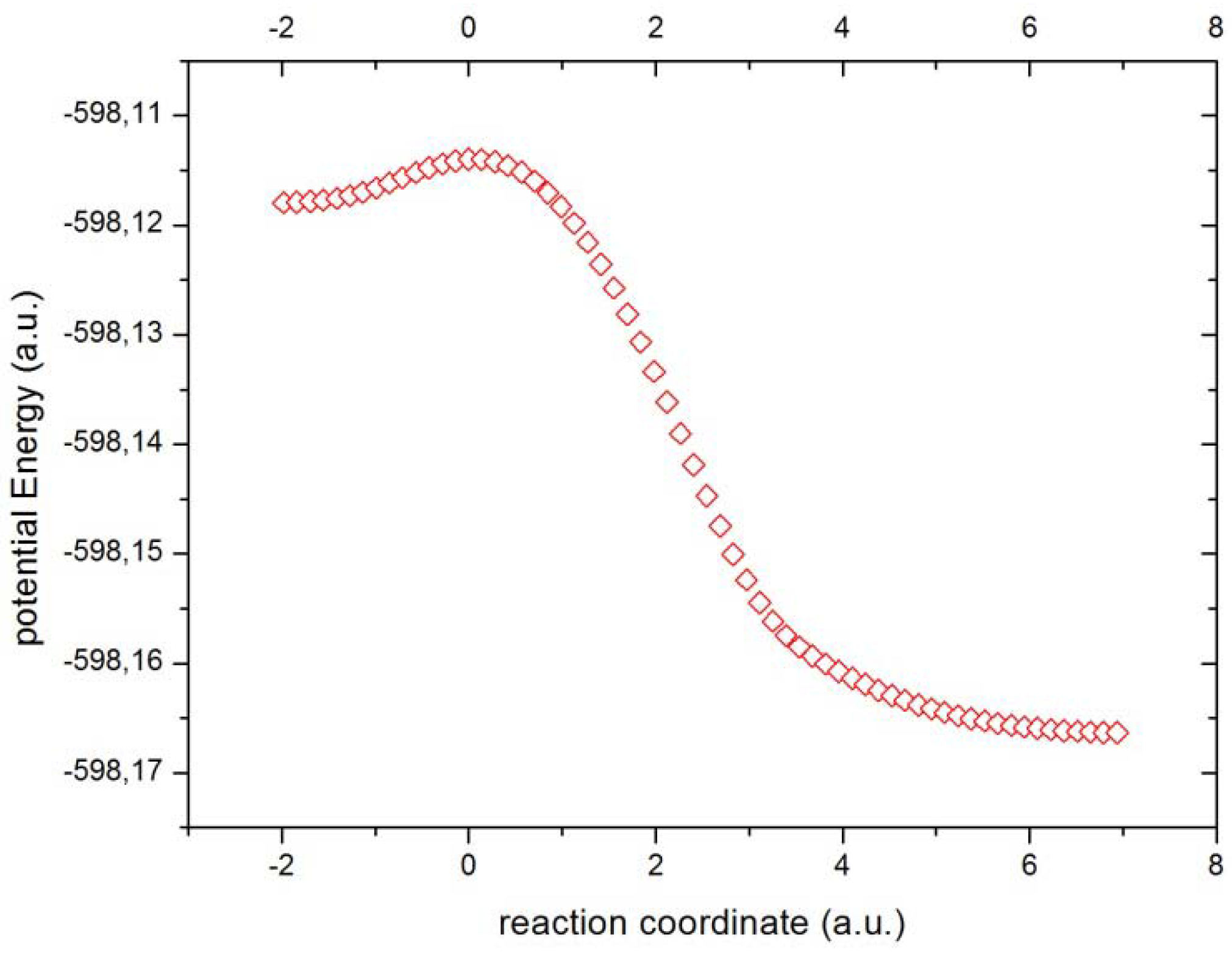
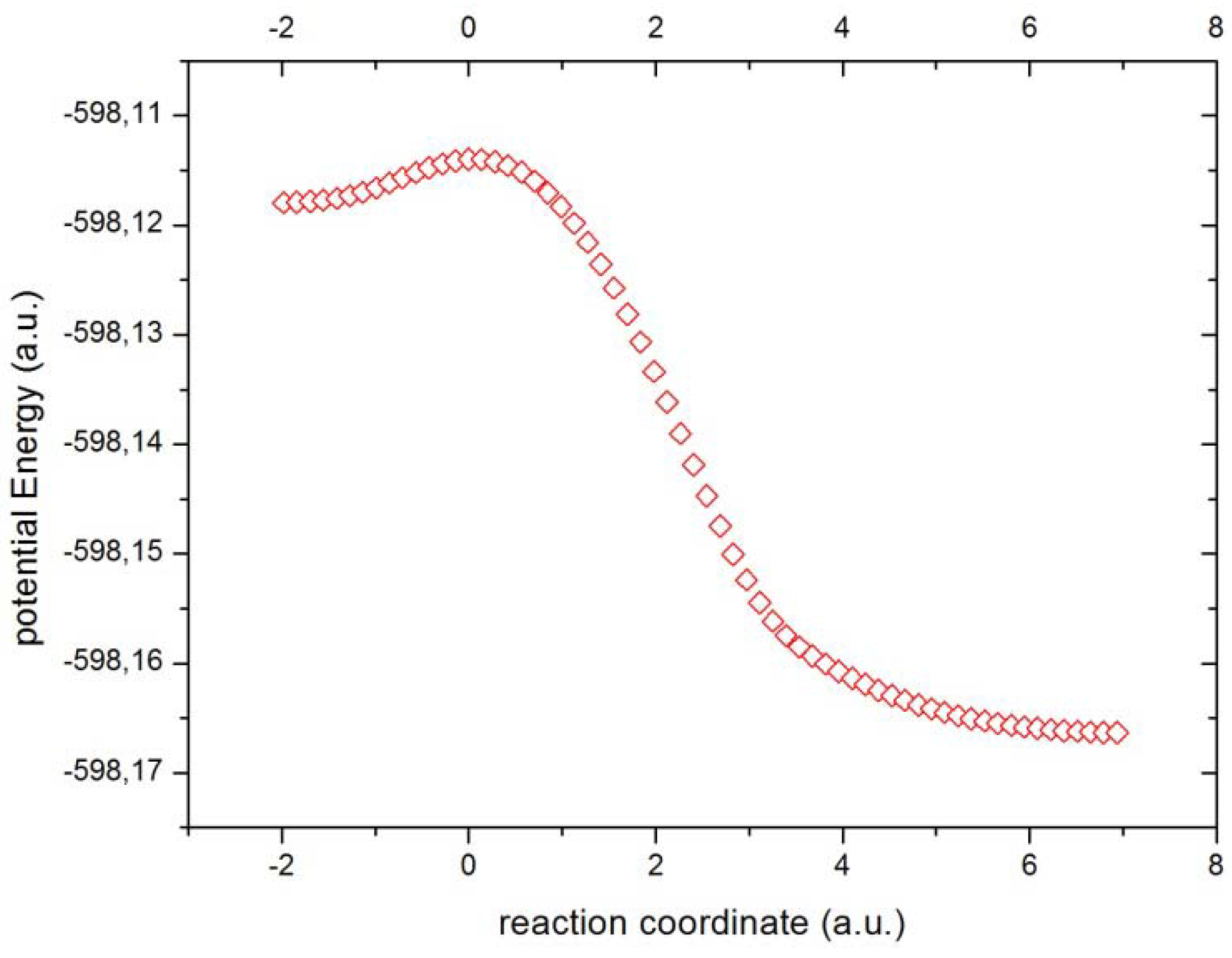
Figure 2 the potential energy surface in mass-weighted coordinates for the internal reaction path (IRC) is depicted. We can observe from
Figure 2 the transition from reactants (higher energy state) to products (lower energy state), passing across the TS which represents an energy barrier and this is perhaps the only important feature which is accurately described by the energy profile. However, there are chemical processes that remain unraveled from the energetic profile. So it happens that these chemical processes are associated with important concurrent phenomena which are phenomenologically described with information-theoretic measures which we are about to employ for the analysis.
Figure 2.
The potential energy (a.u.) through the reaction cordinate (a.u.).
Figure 2.
The potential energy (a.u.) through the reaction cordinate (a.u.).
3.1. Phenomenological Behavior
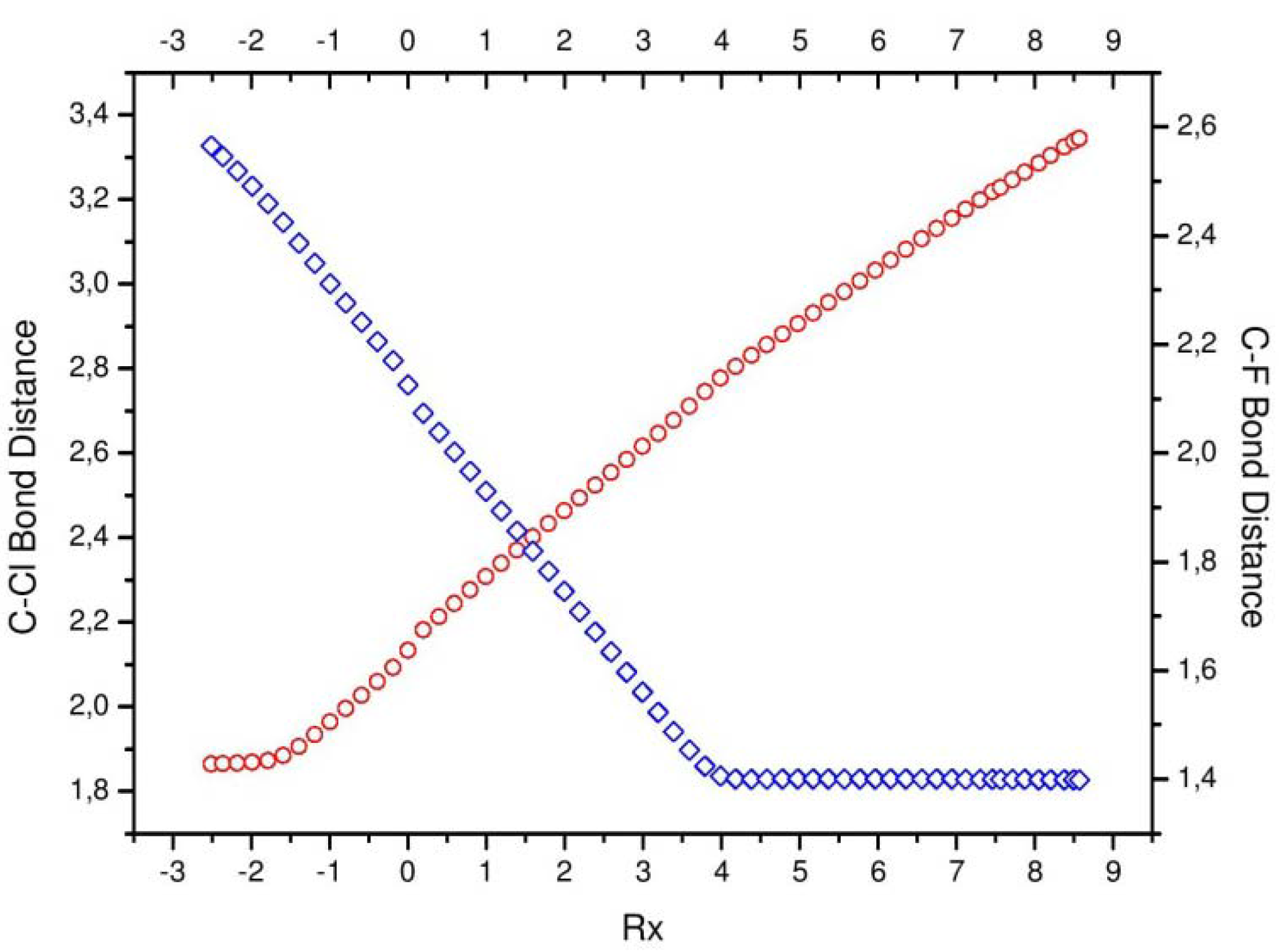
The chemical course of the reaction can be understood physically trough the phenomenological description of some selected physicochemical properties and geometrical parameters. For instance, the bond distances (in a.u.) for the
and the
complexes are determined along the IRC and shown in
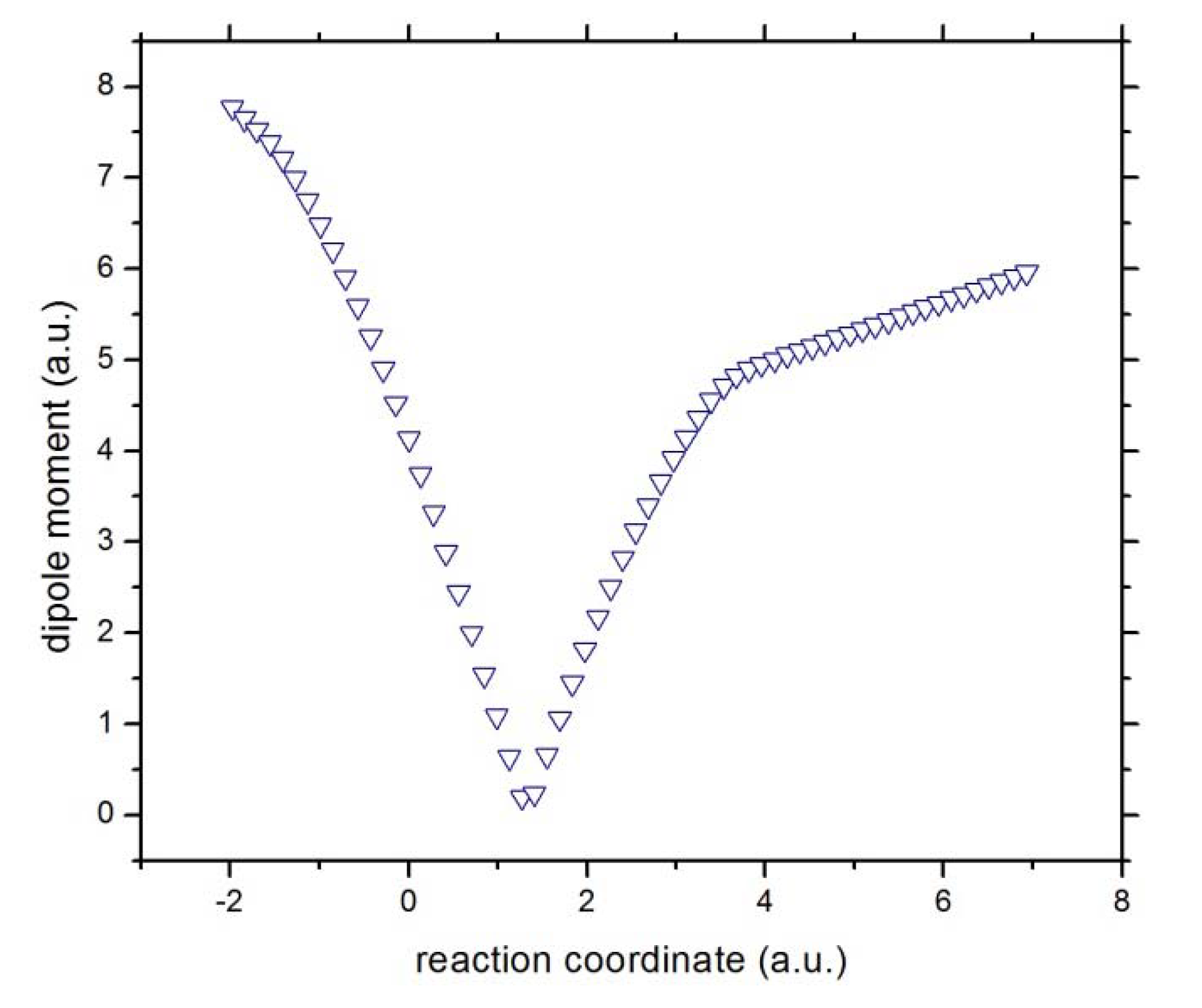
Figure 3. The total dipole moment for the molecular complex is sketched in
Figure 4. The atomic charges for the atomic species of F- and Cl- along with the methyl complex are drawn in
Figure 5. The electric atomic potentials of the methyl complex and the atomic species are outlined in
Figure 6.
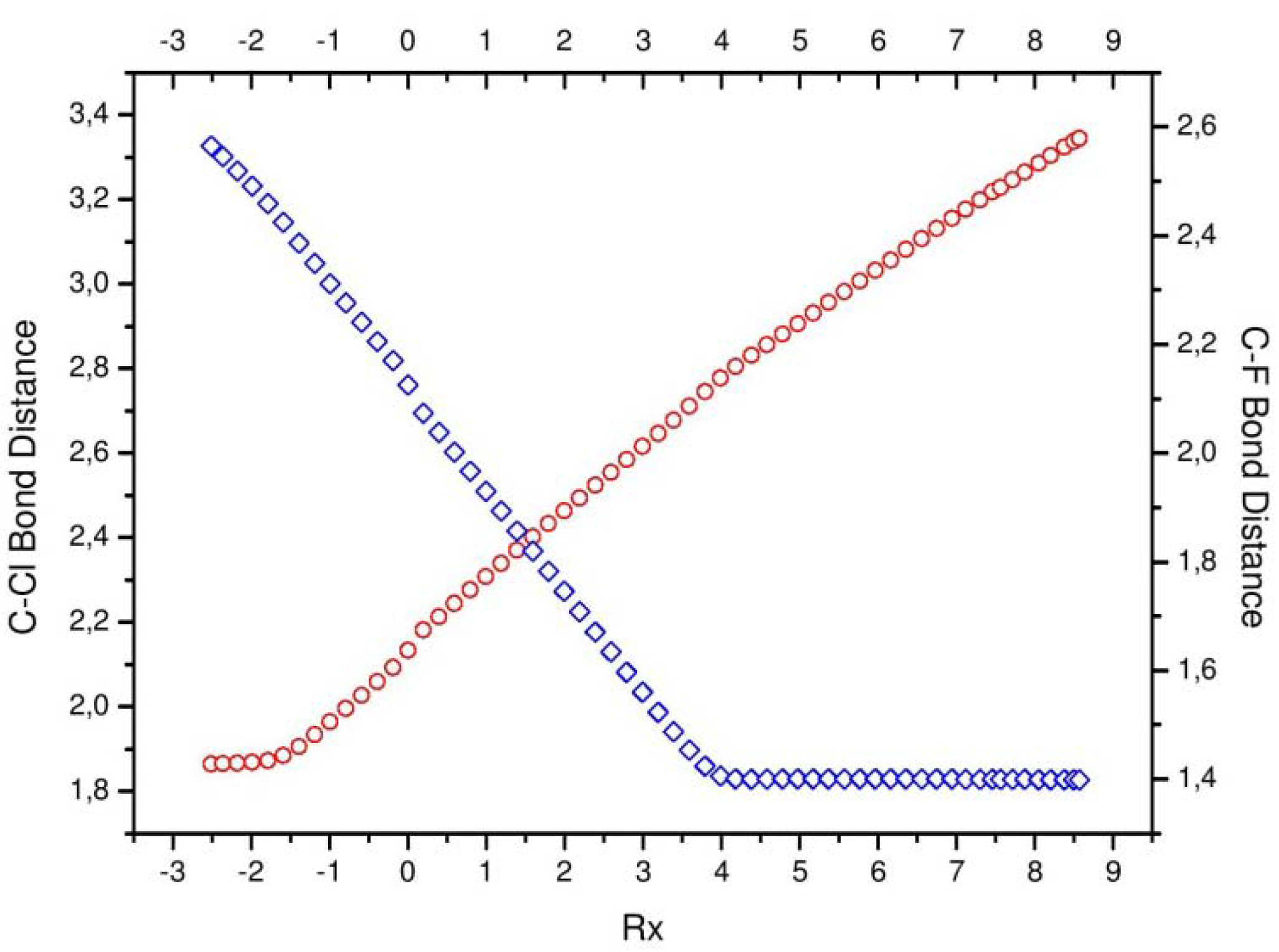
Figure 3.
Bond distances of the [Cl···CH3]* complex (in red) and the [CH3···F]* (in blue).
Figure 3.
Bond distances of the [Cl···CH3]* complex (in red) and the [CH3···F]* (in blue).
The most evident signaling of the concurrent processes happening in the chemical reaction comes from the bond distances (
Figure 3), which show two important moments: at R
x ~ −1 where the
bond starts breaking (bond cleavage region, BCR), and at R
x ~ −4 where the
starts forming (bond forming region, BFR). The above structural signaling concurs with somewhat more intricate processes, which are concurrent in the sense of being interpreted through different physical properties which describe common phenomena.
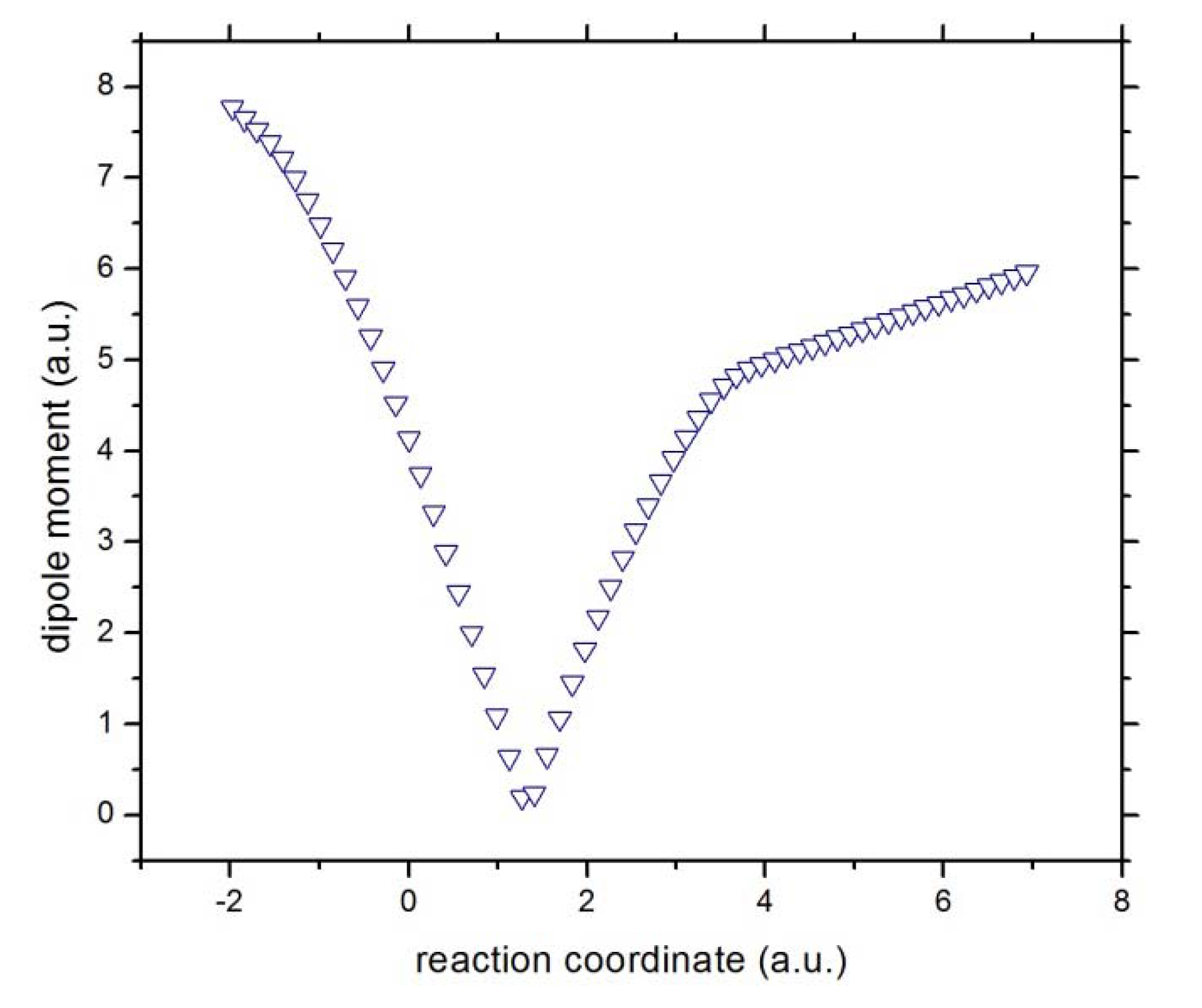
Figure 4.
The total dipole moment of the reactive complex [Cl···CH3···F]−.
Figure 4.
The total dipole moment of the reactive complex [Cl···CH3···F]−.
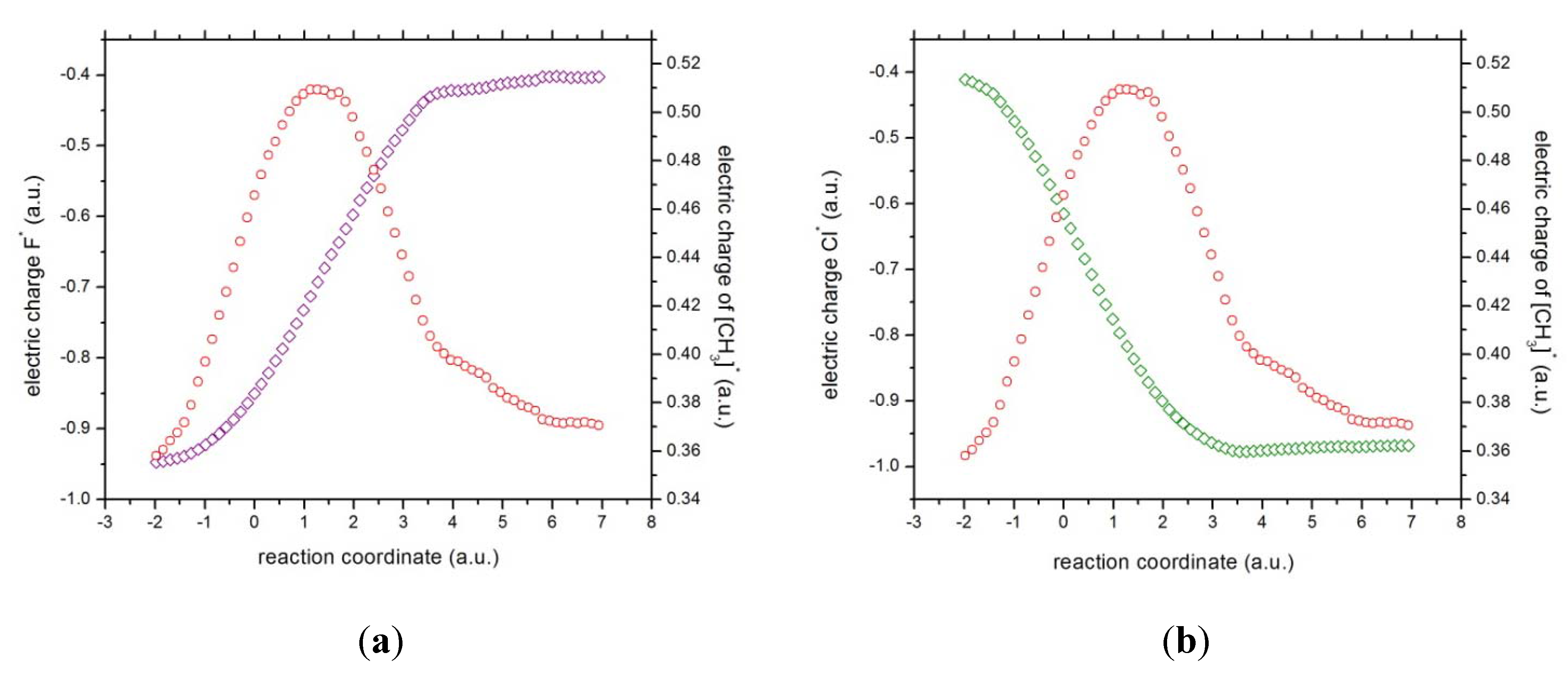
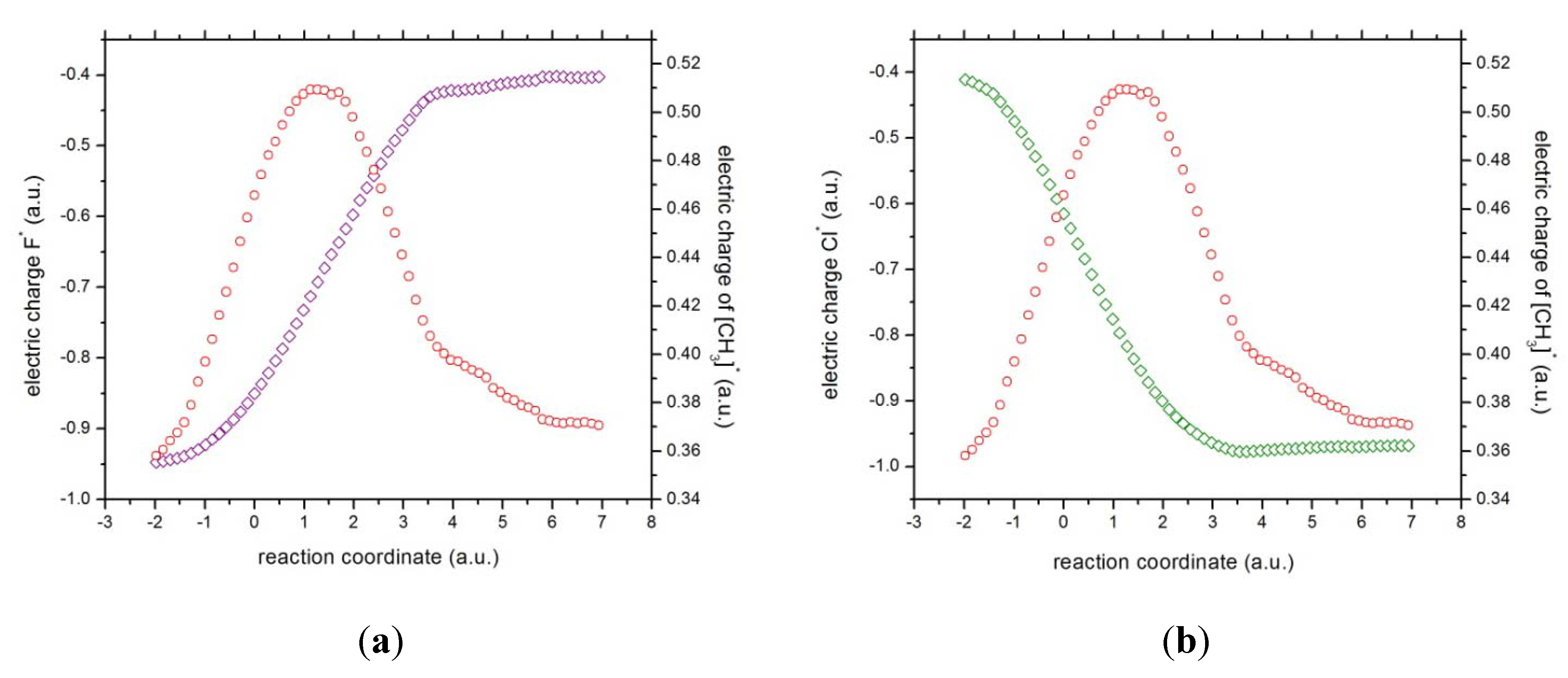
Concurrent processes: we can witness a charge transfer process from fluorine to chloride through the methyl ionic-complex just right from the beginning of the reaction, from R
x ~ −1 to +3, because of the larger electronegativity of fluorine as compared to chlorine. This might be observed from
Figure 5, where the atomic and molecular charges are depicted. The general feature is that as the fluorine transfers charge to the chlorine atom, the methyl complex acts as a buffer,
i.e., as atomic fluorine approaches the molecular complex from R
x ~ −1 to +1 (becoming more positive), the methyl group donates charge to the chlorine atom up to a point where its electrophilic power becomes maximal at Rx ~ +1. Note that at this point the total dipole moment of the whole system becomes null (see
Figure 4),
i.e., the density of the reactive complex
is completely delocalized through the whole complex system (see above). This charge exchange between fluorine and chlorine continues from R
x ~ +1 to +3 (see
Figure 5) and the buffer acts in the opposite way,
i.e., the methyl group gains charge from the fluorine atom up to a point where the atomic and molecular species repel each other at the onset of R
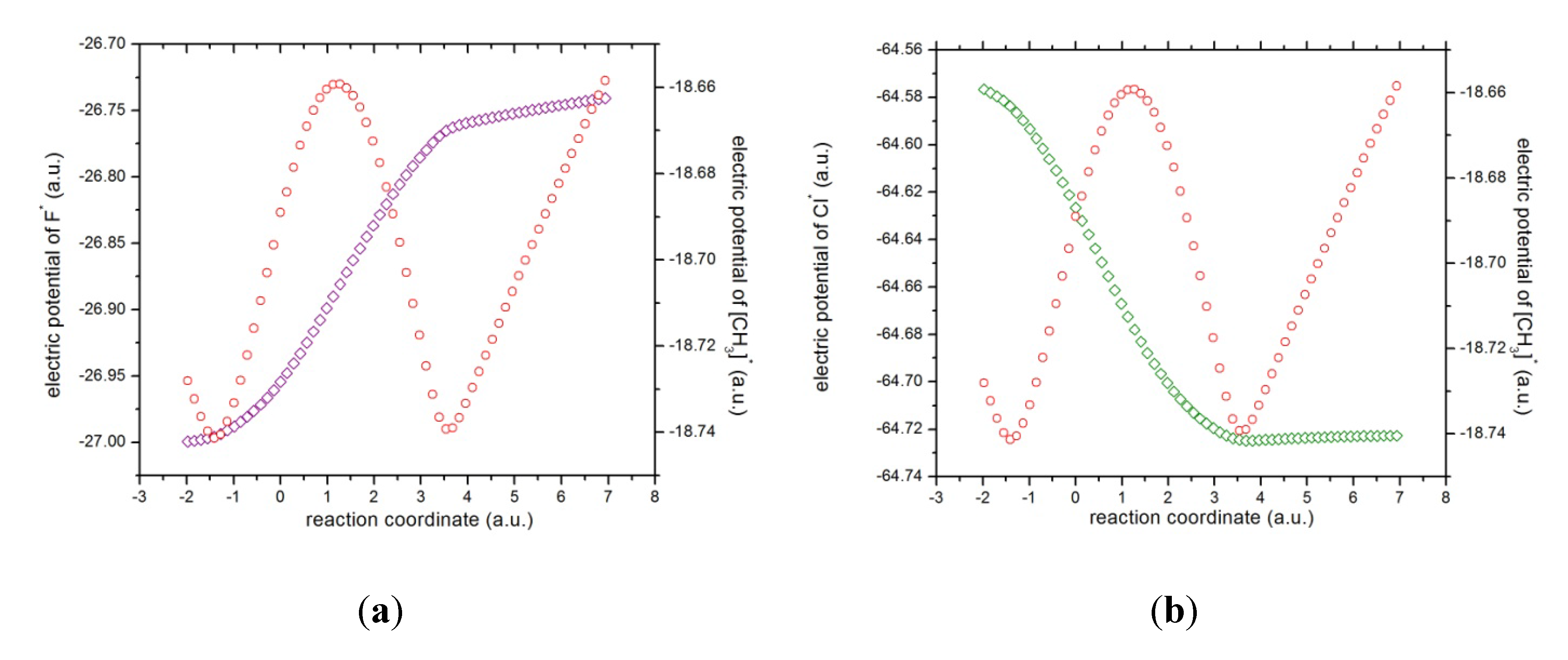
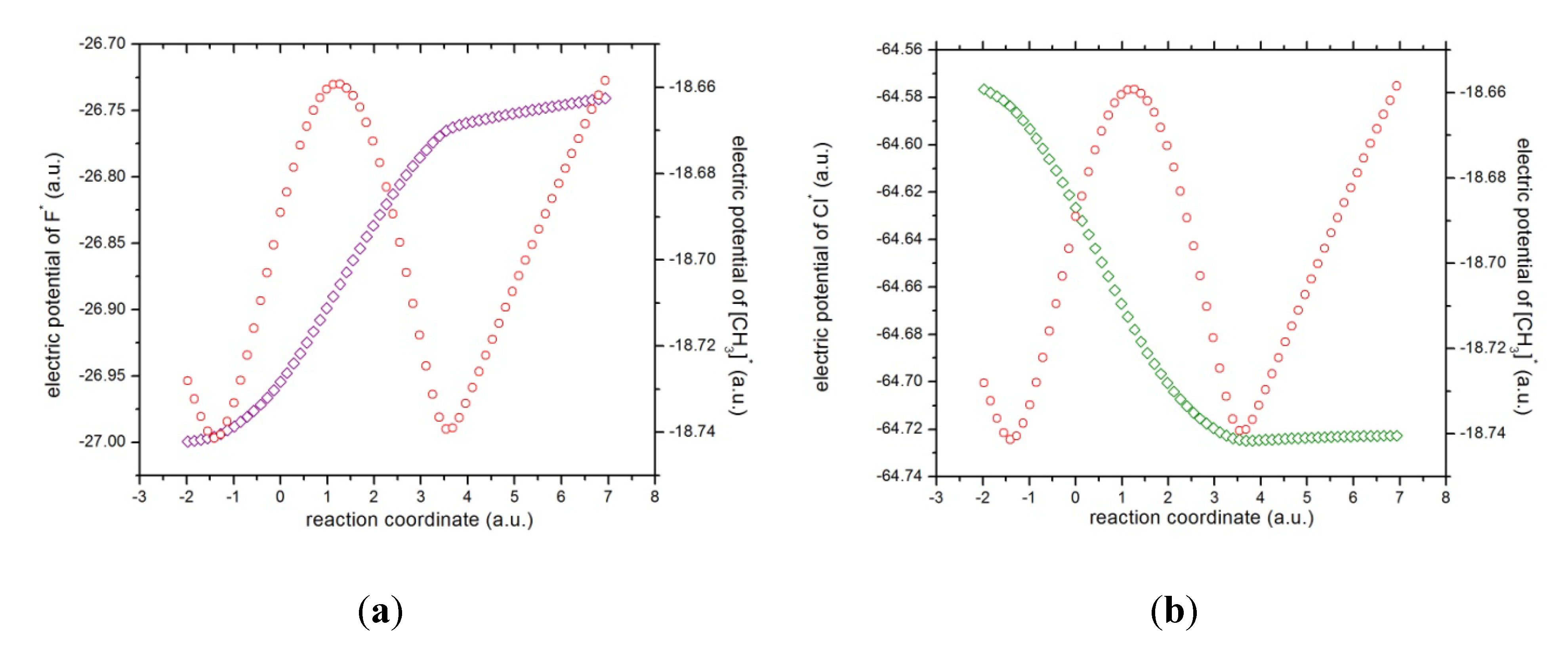
x ~ 4.0. The latter might be verified through the electric potentials between the complex [H
3···F]
* and the chlorine atom, which have been depicted in
Figure 6, from which it is verified that the electric potentials for both species tend to increase,
i.e., the molecular complex
and the atomic chlorine repel each other. It is interesting to note from the atomic potentials (see
Figure 6) that when the reaction initiates, the fluorine atom and the methyl group tend to increase their potentials whereas the chlorine atom acts in the opposite manner up to R
x ~ +1, revealing the attractive forces driving the reaction complex up to R
x ~ +4 where the opposite is observed,
i.e., the electric potentials among the species head in the same direction and hence the repulsive forces act upon as we explain above.
According to the phenomenological behavior the reaction can be described as follows: (i) from the reactants region up to Rx ~ −1 the fluorine atom transfers charge to the molecular complex while the methylic group acts as a buffer and the bond cleavage with chlorine occurs, then (ii) from Rx ~ −1 to the transition state no concurrent phenomena can be associated with it, (iii) from the last point up to Rx ~ +1, charge transferring continues until the molecule stabilizes electrostatically reaching a null dipole moment, (iv) then from Rx ~ +3 to +4 the process inverts and the buffer accepts charge from fluorine up to the bond forming between these two species occurs, and (v) from Rx ~ +4 up to the end of the reaction, the whole process gets inverted (the electric potentials of the products increase) and repulsive forces between the [CH3···F]* and Cl− occur.
Figure 5.
The charges of (a) fluorine (in magenta) and the methyl complex (in red), and (b) chlorine (in green) and the methyl complex (in red).
Figure 5.
The charges of (a) fluorine (in magenta) and the methyl complex (in red), and (b) chlorine (in green) and the methyl complex (in red).
Figure 6.
The electric atomic potentials of (a) fluorine (in magenta) and the methyl complex (in red), and (b) chlorine (in green).
Figure 6.
The electric atomic potentials of (a) fluorine (in magenta) and the methyl complex (in red), and (b) chlorine (in green).
3.2. Shannon Entropy in Conjugated Spaces
The values for the Shannon entropies in position and momentum spaces along the IRC of the reaction are depicted in
Figure 7a,b, respectively.
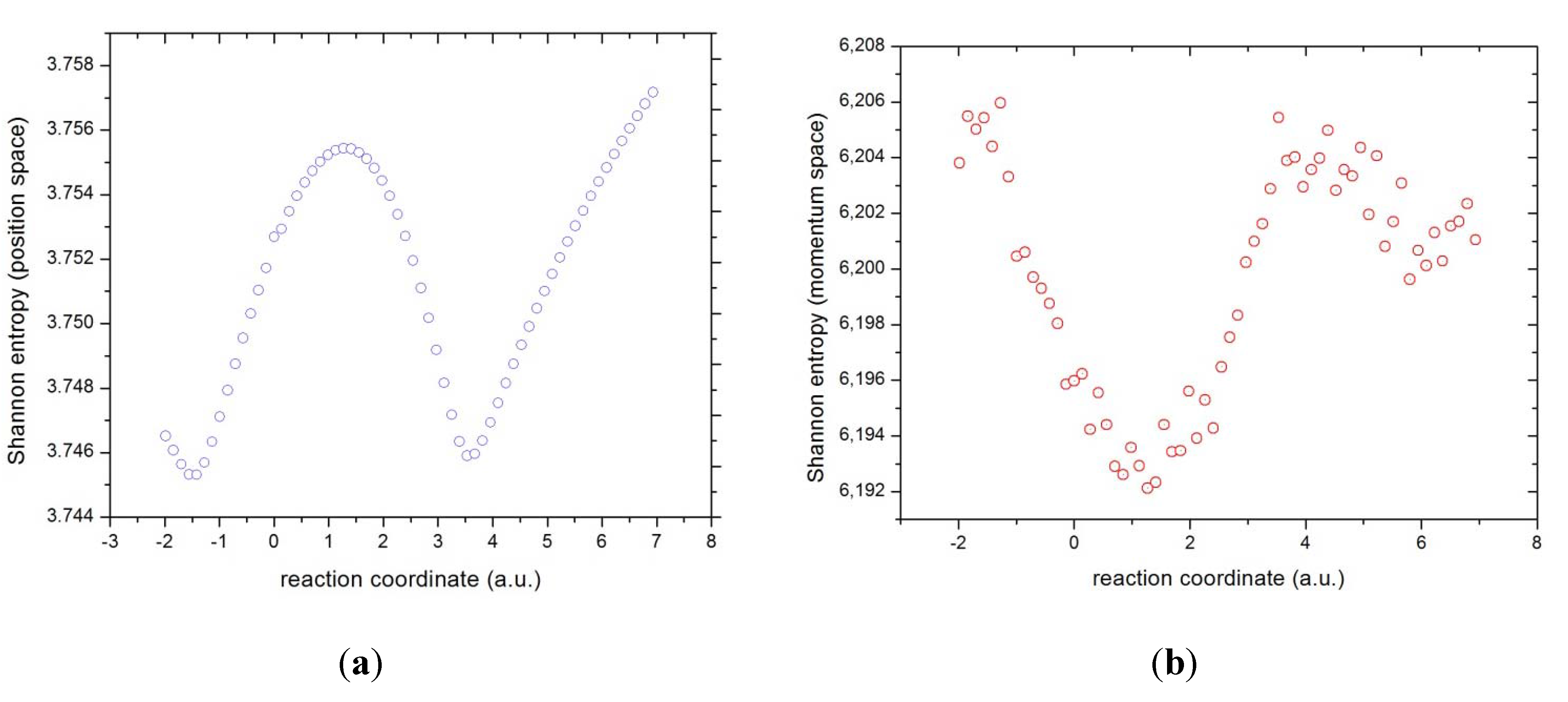
Figure 7.
Shannon entropies in (a) position and (b) momentum spaces for the IRC of the reaction.
Figure 7.
Shannon entropies in (a) position and (b) momentum spaces for the IRC of the reaction.
It has been discussed above that the Shannon entropy is capable of measuring the deviation from localizability to describe new reactivity features on the path of chemical reactions that are not present from the energy profile [
65,
66]. First, we proceed to analyze the electronic densities in either space along the IRC of the chemical reaction in terms of features of localizability/delocalizability and then we associate these features to the phenomenological description of the reaction (see above). Consequently, from
Figure 7a the general aspects described by the Shannon entropy in position space could be summarized as follows: as the reaction initiates, in a region delimitated by R
x ~ −1, a highly localized electronic density for the molecular complex in position space is observed (represented through the electron distributions along the IRC) which signals, according to our analysis of previous subsection, the BCR among chlorine and the methyl group (see
Figure 4). Then as the reaction progresses a highly delocalized density is observed at R
x ~ +1, where the total dipole moment of the molecule is null (see
Figure 5), signaling a non-polarized density. This occurs where the methyl group reaches its minimal charge and its maximum capacity to acquire it (electric potential is maximal). There again, towards the end of the reaction at R
x ~ +4, a highly localized electronic density in position space is observed which indicates the BFR among the fluorine and the methyl group. Aside of these concurrent processes, it is worthy to mention that at R
x = 0, the Shannon entropy is capable of detecting the TS in the form of a slight inflection point despite of the fact that none of the concurrent processes analyzed here seem to be associated with it. Here again, becomes appropriate to quote Shaik in the context of the discussion presented in the introduction section: “notwithstanding that critical points of the energy surface are useful mathematical features for analyzing the reaction-path, their chemical and physical meaning remains uncertain” [
21]. Apparently, for the test reaction analyzed in this work the TS represents a single point at the IRC with little chemical significance, other than the height of the energy barrier of course.
The analysis of the Shannon entropy in momentum space proceeds in analogous way as the position space one, with significant differences though. As discussed earlier, momentum densities are obtained through momentals calculated by means of Fourier transformation of the position space orbitals (see Equation (1), as such is heavily affected by the outer regions of , and hence is highly sensitive to even small geometry changes on the chemical structures at the IRC. Another difference of the momentum density with respect to its position counterpart is that the former represents the kinetic energy content of the distributions and hence the information content is explained in different physical terms: a lower Shannon entropy in this space represents a more localized momentum density, which in turn reflects a sharper region in the distribution of electron velocities corresponding with a lower kinetic energy region. In contrast, a higher Shannon entropy in momentum space describes a more delocalized momentum density, reflecting a broader region of electron velocities and hence a higher kinetic energy.
As a consequence of the above position space densities should include diffuse functions in the orbital base representation to adequately describe the momentals. On the other hand the numerical integrations necessary to compute the information measures (
S,
I, or
D) may require very tight error thresholds; usually, for the position space properties a threshold of 10
−5 is sufficient to produce very smooth functions like the ones shown in
Figure 7a (or the ones for
I and
D shown below). However, in order to obtain similar quality functions, for instance the Shannon entropy in momentum space shown in
Figure 7b, we had to recur to very demanding integrations with numerical errors of the order of 10–10, and still some numerical noise remains especially in the Shannon entropy. Even though, clear trends are observed from
Figure 7b,
i.e., a decreasing behavior of the Shannon entropy at the beginning of the reaction up to R
x ~ +1, indicating that the reactants (represented through the electron distributions along the IRC) initiate the reaction with a higher kinetic energy which diminishes as the reaction progresses until the molecular complex holds a null dipole moment (a delocalized momentum density which turns to become localized). This is in agreement with discussion above in that the impulse of the reaction is originated from the electrostatic attractive interaction between the reactants in the first stage which causes the transferring of charge from fluorine and the buffer (methylic group) to the chlorine atom, ending at R
x ~ +1 where the whole molecule stabilizes by charge equalization to nullify the total dipole moment. This is witnessed by the methylic group through a maximal deficiency of charge (
Figure 5) and maximal electric potential (
Figure 6), as discussed above. In the second stage, the opposite behavior occurs, the flow of charge goes from the fluorine to the methylic group and chlorine. This again is witnessed by the methylic group which exerts the inverse phenomenon so as to acquire maximal accumulation of charge and electric potential at R
x ~ +4, where the bond between the fluorine and the methyl group is formed. Once more, the agreement with the behavior of the momentum space Shannon entropy is observed by the increase of the kinetic energy in this region, from R
x ~ +1 to +4, which is employed for bond forming. Finally, in the last stage the whole process gets reversed,
i.e., repulsive electrostatic interactions arise once the products of the reaction are formed because of the increasing electric potentials. This is revealed by the Shannon entropy which reflects the stabilization of the whole system through the lowering of the total energy indicating the end of the chemical process.
3.3. Information Planes
In the search of any joint features of uniformity-localizability (D-L), and disorder-localizability (I-J) for the chemical course of the reaction we have found useful to plot the contribution D and L to the total LMC complexity, and similarly with I and J to the FS complexity. Note that all information products that we are studying are good candidates to form complexity measures, i.e., to preserve the desirable properties of invariance under scaling, translation and replication.
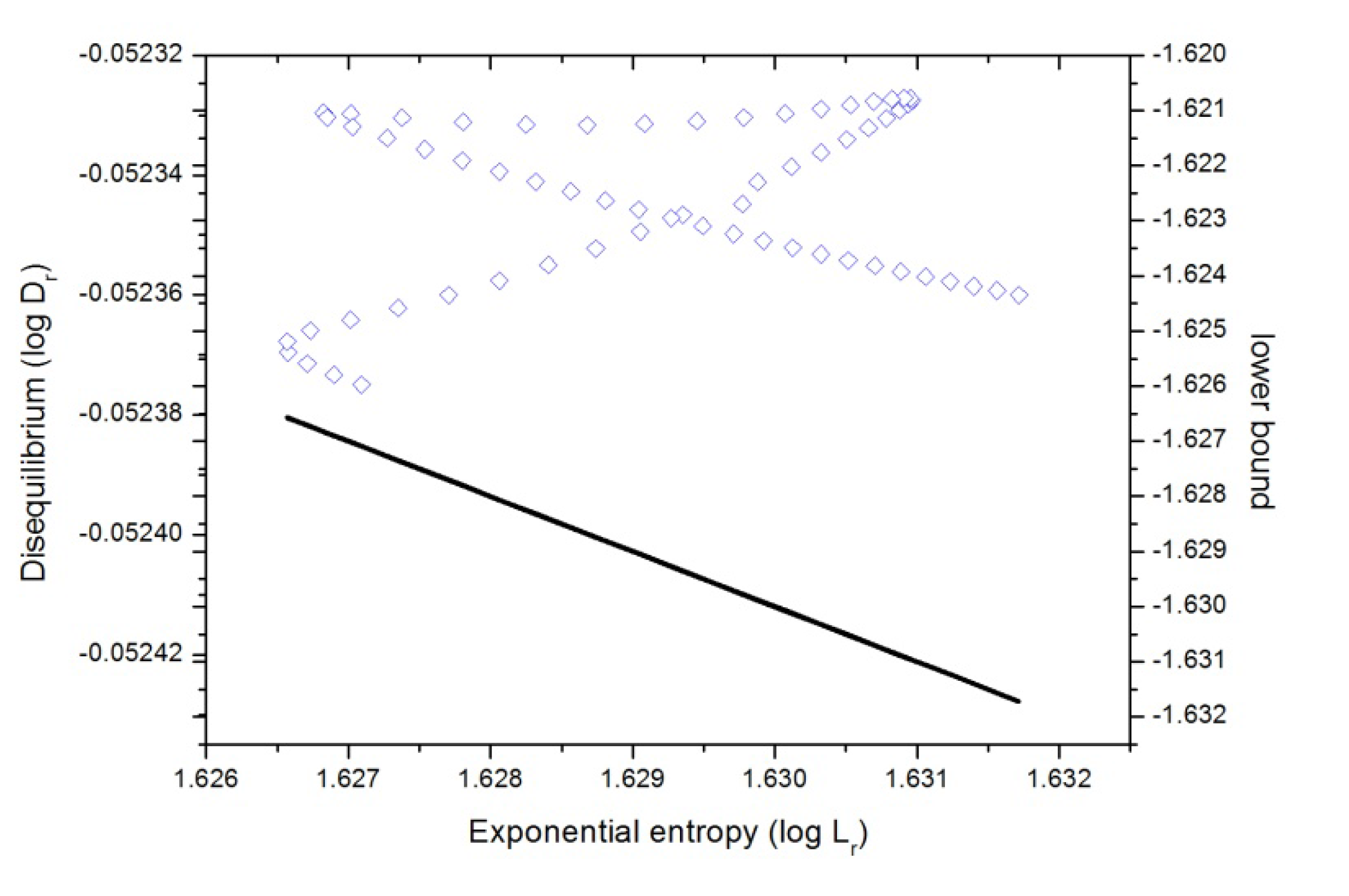
We have depicted in
Figure 8 the
D-L plane in position space. At this point it is worth mentioning that there is a rigorous lower bound to the associated
C(LMC) complexity, given by Equation (6), which is
C(LMC) =
D·L ≥ 1 for both spaces. From
Figure 8 we may observe that the
D-L plane is clearly separated into two regions, according to the
inequality (valid for position, momentum as well as product spaces), and the region below the line (equality) corresponds with the forbidden region. Parallel lines to this bound represent isocomplexity regions, showing that an increase (decrease) in
uncertainty,
L, along them is compensated by a proportional decrease (increase) of disequilibrium, and higher deviations from this frontier are associated with greater LMC complexities.
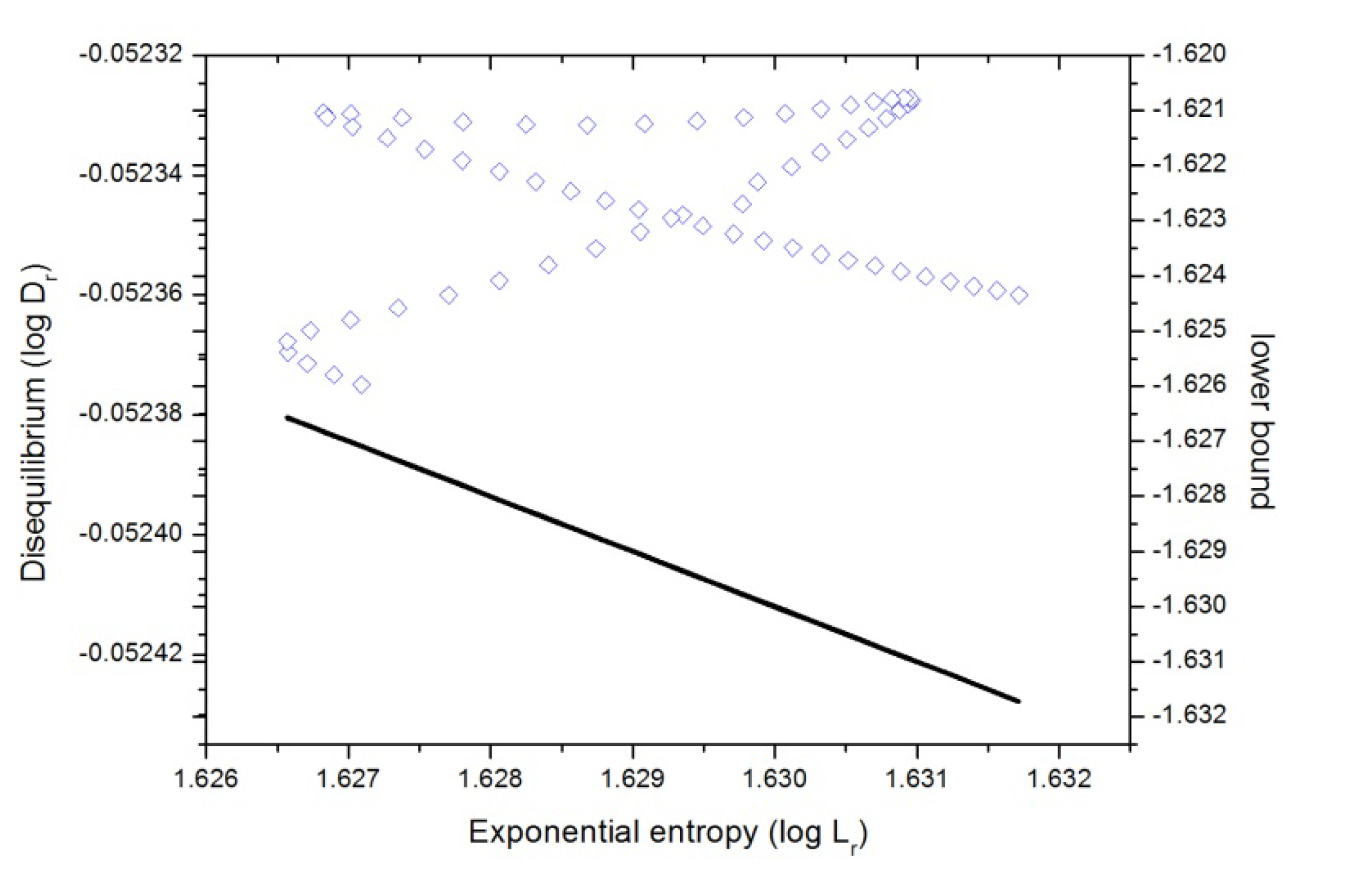
From
Figure 8 we may note the following: from the R (reactants region) to the bond-breaking (
B-B) region for chlorine, at R
x ~ −1, as well as the region from the bond forming (
B-F), at R
x ~ +4, to the P (products), the behavior is isocomplex to the
C(LMC) bound, whereas for the rest of the IRC it behaves in a more complex manner. The general observations are that at the first region (see previous section) the plane shows a minimum
uncertainty (highly localized structure at
B-B) at the expense of loosing
uniformity, then in the next stage where the charge transfer from fluorine to chlorine occurs, uniformity is gained at the expense of the reactants density becoming more delocalized up to the total dipole moment of the molecular complex becomes null at R
x ~ +1. From this point the opposite occurs, the molecular complex gets
localized whereas it slightly augments
uniformity up to the B-F region where the fluorine gets attached at R
x ~ +4. Finally, in the last stage, where the products repel each other, the molecular complex gets more
delocalized at the expense of becoming more
uniform.
Figure 8.
Disequilibrium-Shannon plane (D-L) in position space (in blue) for the IRC of the SN2 reaction in double logarithmic scale, i.e., according to Equations (2) and (6), , where equality stands for the lower bound. This case is also depicted in the figure (black line).
Figure 8.
Disequilibrium-Shannon plane (D-L) in position space (in blue) for the IRC of the SN2 reaction in double logarithmic scale, i.e., according to Equations (2) and (6), , where equality stands for the lower bound. This case is also depicted in the figure (black line).
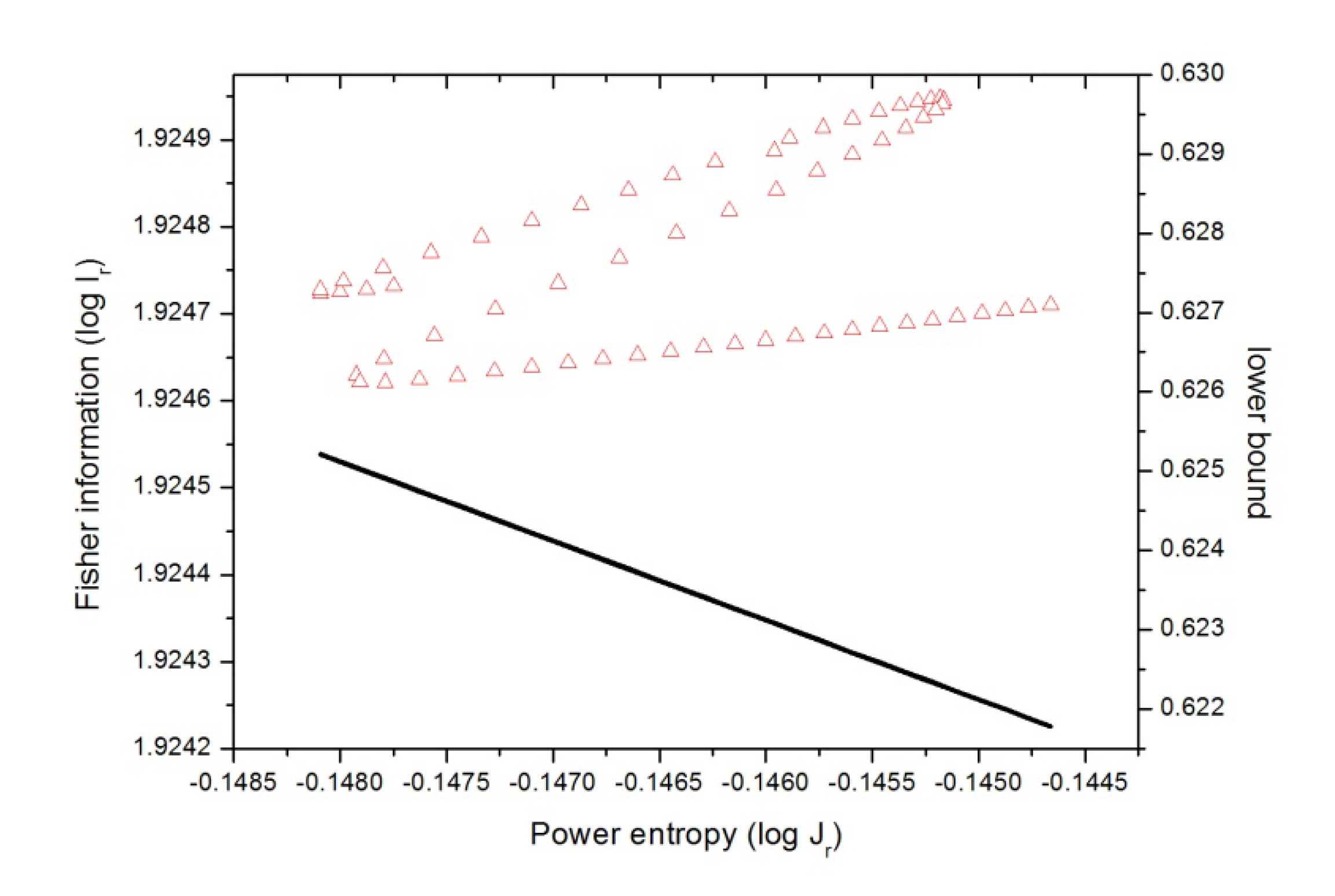
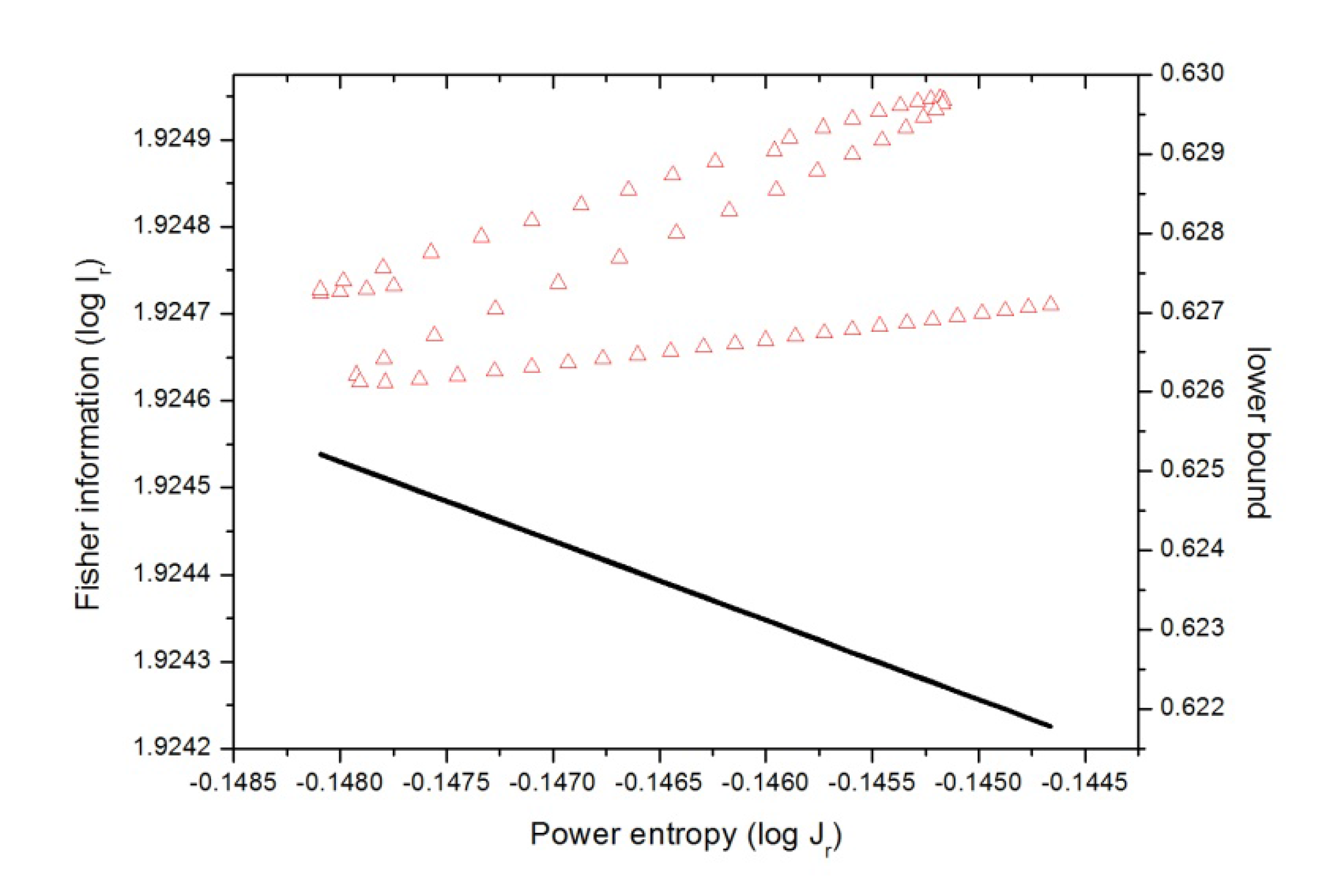
In
Figure 9 we have plotted (in a double-logarithmic scale) the
Ir-Jr plane (position space) for the chemical reaction. At this point it is worth mentioning that there is a rigorous lower bound to the associated
C(FS) complexity, given in Equation (11), which is
C(FS) =
I·J ≥ 3 for both spaces. Thus,
Figure 9 indicates a division of the
Ir-Jr plane into two regions where the straight line
IJ = 3 (drawn in the plane in logarithmic scale) divides it into an “allowed” (upper) and a “forbidden” (lower) part. As it may be observed from
Figure 9 the R region is characterized by getting a
localized molecular complex (position space density) as it becomes
dis
ordered at R
x ~ −1, then when the atomic species exchange charge up to R
x ~ +1 both qualities augment,
order and
uncertainty, and the molecular complex possesses a null dipole moment. Then in the next stage the opposite occurs, both qualities decrease and the molecular complex density gets disordered at the expense of becoming localized as the charge is exchanged up to the B-F region is reached at R
x ~ +4. In the last stage, when the products exert repulsion forces the molecular complex (position space density) becomes delocalized at the expense of gaining order. It is worth noting that none of the concurrent processes as depicted in
Figure 9 are isocomplex to the
I-J plane.
Figure 9.
Fisher-Shannon plane (I-J) in position space (in red) for the IRC of the SN2 reaction in double logarithmic scale, i.e., according to Equations (9) and (11), , where equality stands for the lower bound. This case is also depicted in the figure (black line).
Figure 9.
Fisher-Shannon plane (I-J) in position space (in red) for the IRC of the SN2 reaction in double logarithmic scale, i.e., according to Equations (9) and (11), , where equality stands for the lower bound. This case is also depicted in the figure (black line).
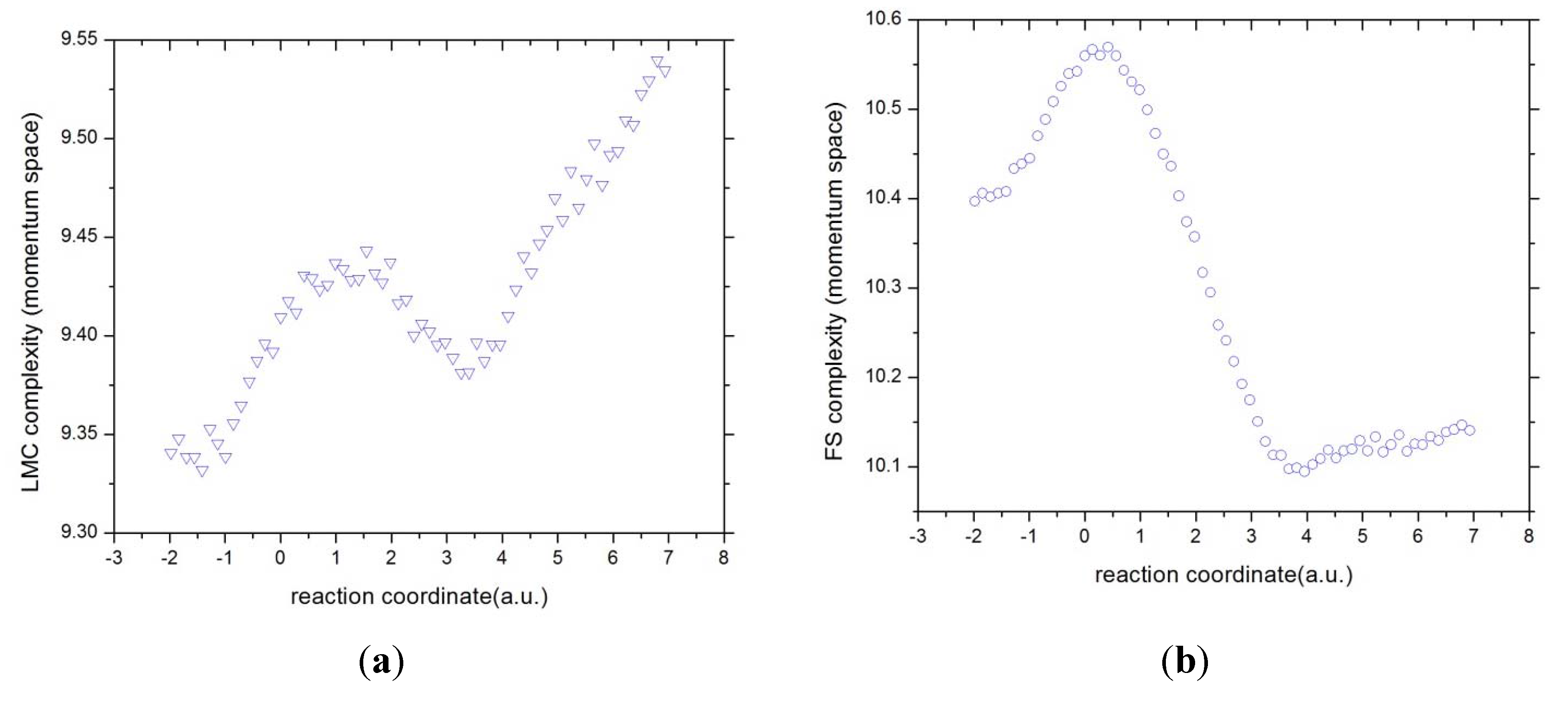
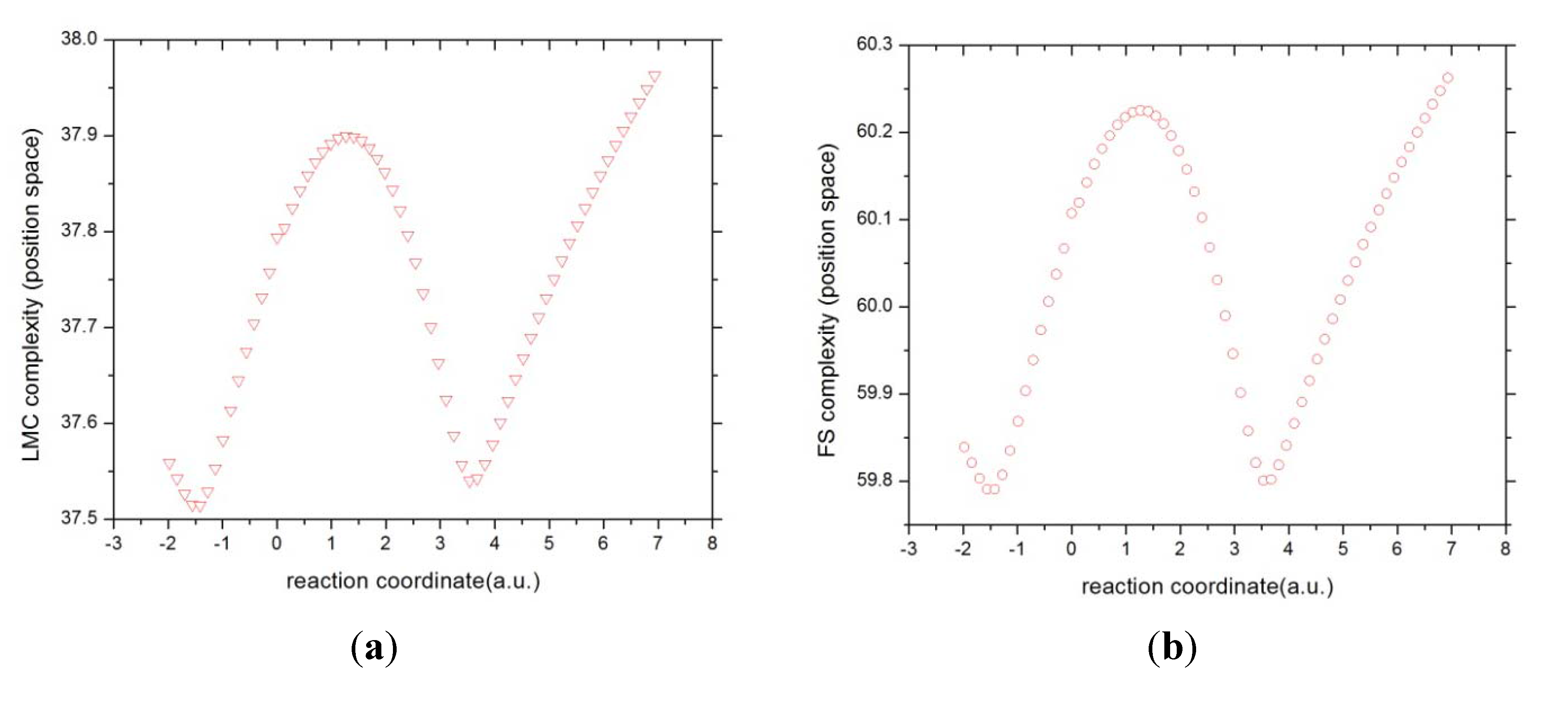
3.4. Complexity Measures
In the search of joint patterns of
uniformity-localizability through
C(LMC) and
disorder-localizability through
C(FS) we have depicted in
Figure 10 and
Figure 11 the values for these complexity measures in position and momentum spaces, respectively. The general observation from
Figure 10 is that both complexity measures behave similarly in position space. It may be observed that the R region hold minimum complexity at the B-B region and as the reaction evolves by the charge exchange between the atomic species, both complexities augment; this happens up to the maximally delocalized state is reached (see above), its chemical implications will be discussed elsewhere. Then, as the exchange of charge continues, the complexities decrease up to the B-F region holds the minimum complexity value at the IRC. Finally, at the final stage where the product species repel each other, both complexities increase. It is worth noting that both reaction moments, B-B and B-F, when the bonds are made and destroy, hold the same complexity behavior. Also interesting is noting that across the study, we could not relate the TS to any specific concurrent process with chemical sense. However, it seems that both complexities are capable of detecting the TS in the manner of a slight inflection point at R
x = 0.
Figure 10.
The complexities in position space (a) C(LMC) and (b) C(FS) for the IRC of the SN2 reaction.
Figure 10.
The complexities in position space (a) C(LMC) and (b) C(FS) for the IRC of the SN2 reaction.
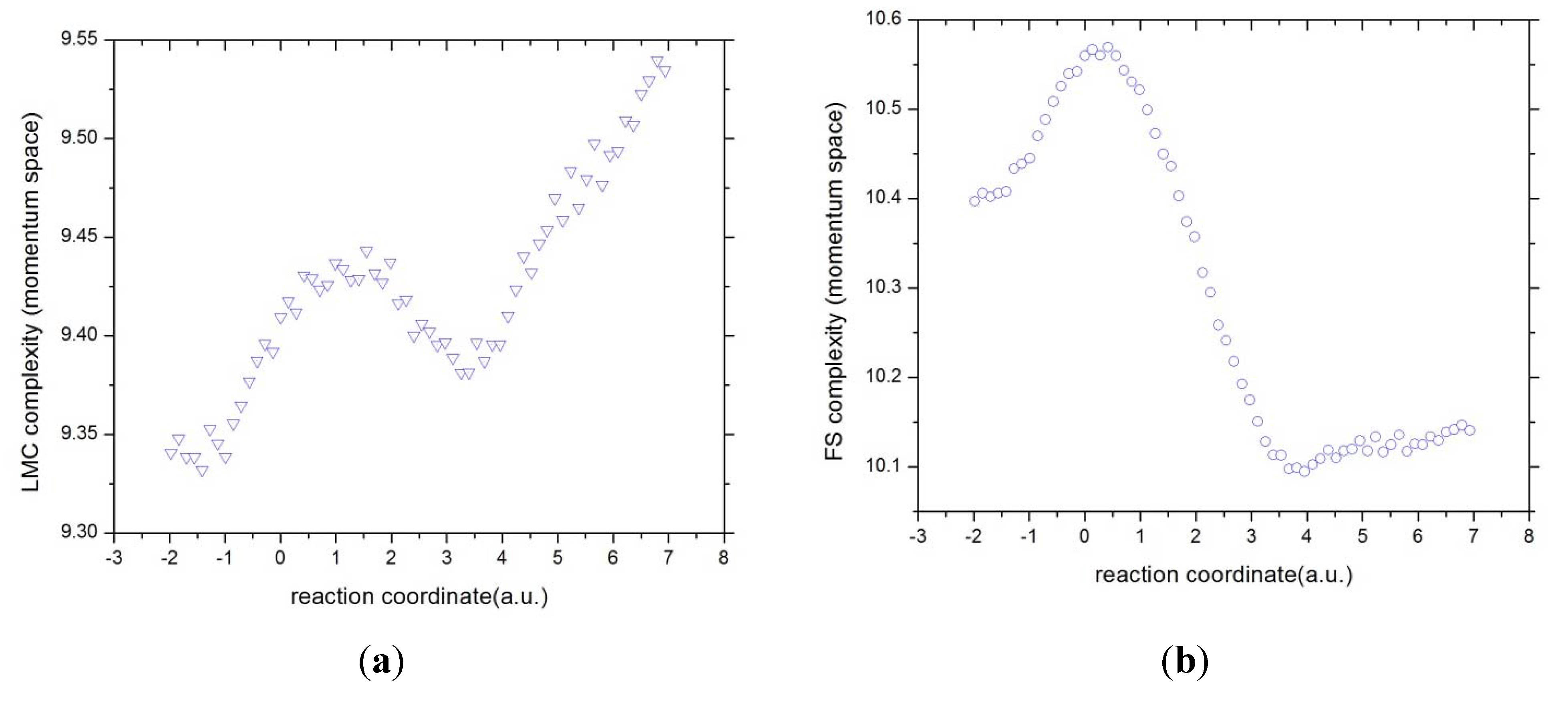
Figure 11.
The complexities in momentum space (a) C(LMC) and (b) C(FS) for the IRC of the SN2 reaction.
Figure 11.
The complexities in momentum space (a) C(LMC) and (b) C(FS) for the IRC of the SN2 reaction.
In momentum space the C(LMC) and the C(FS) measures look different as those in position space. Although it is apparent that both of them are capable of detecting the aforementioned concurrent phenomena, the last stage is featured by an increment of complexity in their uniformity/delocalization aspects whereas the C(FS) is characterized by lowering of their order/uncertainty aspects. Note also that both moments, B-B and B-F, do not possess the same features for any of the complexities in momentum space. Let us remind the reader that momentum space is characterized by the electronic velocity distribution and hence by the electron kinetic energy. So that, apparently, the concurrent processes of the reaction happen at different energy stages.
4. Conclusions
In this work we have investigated the SN2 exchange reaction of CH3Cl + F− by use of information-theoretic functionals S, I and D in conjugated spaces. Besides, information planes I-J, D-L and complexity measures C(LMC) and C(FS) were investigated.
It has been the purpose of the present study to show that all of the theoretical-information measures analyzed here are of chemical interest by recovering the simplest concepts employed in the physicochemical organic chemistry language through processes such as bond forming/breaking, charge transfer/reorganization, and charge repulsion. Note that whereas these chemical concepts have been employed in the chemical literature for centuries, there is no direct connection nor simple description when other energy based physical descriptors are employed. In contrast, information theory concepts such as uncertainty measures, information planes and complexities allow to revealing any of the chemical concepts associated with this reaction. This is achieved by the different aspects of uncertainty/uniformity, order/delocalization that have been discussed at the light of the concurrent phenomena of the reaction: B-B, B-F, charge-exchange, maximum delocalization state, and repulsion. On the other hand, according to the phenomenological analysis we have concluded that the TS (critical point) for the SN2 reaction studied here, does not hold any chemical significance other than the energetic barrier which is useful for kinetic purposes. Indeed, none of the concurrent chemical phenomena indicated by the physical properties (atomic charges, electric potentials, geometric parameters, dipole moment) and the theoretical-information measures [S, I, D, L-D, F-J, C(FS), C(LMC)] seem be associated with the TS indicated by the first order saddle of maximum energy. However, point with the TS remains as a quest for physical organic chemistry and our results corroborates this statement. The majority of our information-theoretic measures did not find any chemically significant process linked to the TS. Further, we have characterized a new state in the transition region that seems to possess important chemical information, which corresponds to a maximum delocalized state. This observation indicates that further investigations are necessary in order to improve our knowledge about the transient zone and the chemical phenomena occurring in that region, and also the existence of a maximum delocalized state.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}