
Interplay between Lattice Distortions, Vibrations and Phase Stability in NbMoTaW High Entropy Alloys

Abstract
:1. Introduction
2. Materials and Methods
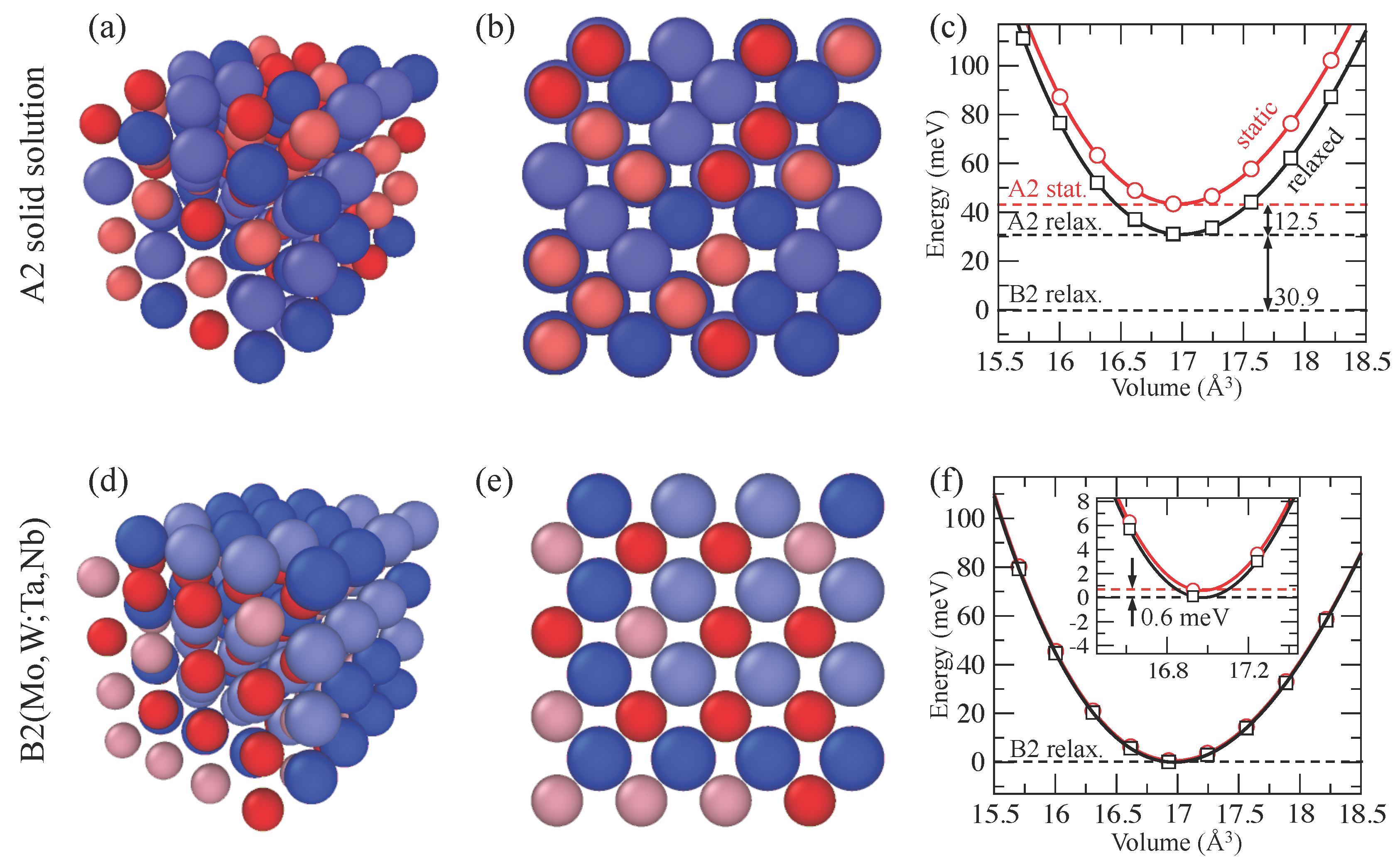
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yao, H.; Qiao, J.W.; Gao, M.C.; Hawk, J.A.; Ma, S.G.; Zhou, H. MoNbTaV medium-entropy alloy. Entropy 2016, 18, 189. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, H.; Spolenak, R. Ultrastrong ductile and stable high-entropy alloys at small scales. Nat. Commun. 2015, 6, 7748. [Google Scholar] [CrossRef] [PubMed]
- Senkov, O.N.; Wilks, G.B.; Miracle, D.B.; Chuang, C.P.; Liaw, P.K. Refractory high-entropy alloys. Intermetallics 2010, 18, 1758–1765. [Google Scholar] [CrossRef]
- Senkov, O.N.; Wilks, G.B.; Scott, J.M.; Miracle, D.B. Mechanical properties of Nb25Mo25Ta25W25 and V20Nb20Mo 20Ta20W20 refractory high entropy alloys. Intermetallics 2011, 19, 698–706. [Google Scholar] [CrossRef]
- Maiti, S.; Steurer, W. Structural-disorder and its effect on mechanical properties in single-phase TaNbHfZr high-entropy alloy. Acta Mater. 2016, 106, 87–97. [Google Scholar] [CrossRef]
- Zou, Y.; Maiti, S.; Steurer, W.; Spolenak, R. Size-dependent plasticity in an Nb25Mo25Ta25W25 refractory high-entropy alloy. Acta Mater. 2014, 65, 85–97. [Google Scholar] [CrossRef]
- Murty, B.S.; Yeh, J.W.; Ranganathan, S. High-Entropy Alloys; Butterworth-Heinemann: Oxford, UK, 2014. [Google Scholar]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Gao, M.C.; Yeh, J.W.; Liaw, P.K.; Zhang, Y. High-Entropy Alloys: Fundamentals and Applications, 1st ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Huhn, W.P. Thermodynamics from First Principles: Prediction of Phase Diagrams and Materials Properties Using Density Functional Theory. Ph.D. Thesis, Carnegie Mellon University, Pittsburgh, PA, USA, May 2014. [Google Scholar]
- Huhn, W.P.; Widom, M. Prediction of A2 to B2 phase transition in the high-entropy alloy Mo-Nb-Ta-W. JOM 2013, 65, 1772–1779. [Google Scholar] [CrossRef]
- Widom, M.; Huhn, W.P.; Maiti, S.; Steurer, W. Hybrid monte carlo/molecular dynamics simulation of a refractory metal high entropy alloy. Metall. Mater. Trans. A 2014, 45, 196–200. [Google Scholar] [CrossRef]
- Del Grosso, M.F.; Bozzolo, G.; Mosca, H.O. Determination of the transition to the high entropy regime for alloys of refractory elements. J. Alloy. Compd. 2012, 534, 25–31. [Google Scholar] [CrossRef]
- Del Grosso, M.; Bozzolo, G.; Mosca, H.O. Modeling of high entropy alloys of refractory elements. Physica B 2012, 407, 3285–3287. [Google Scholar] [CrossRef]
- Körmann, F.; Ruban, A.V.; Sluiter, M.H. Long-ranged interactions in bcc NbMoTaW high-entropy alloys. Mater. Res. Lett. 2016, 0, 1–6. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Zunger, A.; Wei, S.H.; Ferreira, L.G.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.C.; Niu, C.; Jiang, C.; Irving, D.L. Applications of Special Quasi-random Structures to High-Entropy Alloys. In High-Entropy Alloys; Springer: Cham, Switzerland, 2016; pp. 333–368. [Google Scholar]
- Vinet, P.; Ferrante, J.; Rose, J.H.; Smith, J.R. Compressibility of solids. J. Geophys. Res. Solid Earth 1987, 92, 9319–9325. [Google Scholar] [CrossRef]
- Moruzzi, V.L.; Janak, J.F.; Schwarz, K. Calculated thermal properties of metals. Phys. Rev. B 1988, 37, 790. [Google Scholar] [CrossRef]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Oh, H.S.; Ma, D.; Leyson, G.P.; Grabowski, B.; Park, E.S.; Körmann, F.; Raabe, D. Lattice distortions in the FeCoCrNiMn high entropy alloy studied by theory and experiment. Entropy 2016. submitted. [Google Scholar]
- Takizawa, S.; Terakura, K.; Mohri, T. Electronic theory for phase stability of nine AB binary alloys, with A = Ni, Pd, or Pt and B = Cu, Ag, or Au. Phys. Rev. B 1989, 39, 5792. [Google Scholar] [CrossRef]
- Laks, D.B.; Ferreira, L.G.; Froyen, S.; Zunger, A. Efficient cluster expansion for substitutional systems. Phys. Rev. B 1992, 46, 12587. [Google Scholar] [CrossRef]
- Lu, Z.; Wei, S.H.; Zunger, A.; Frota-Pessoa, S.; Ferreira, L.G. First-principles statistical mechanics of structural stability of intermetallic compounds. Phys. Rev. B 1991, 44, 512. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Toda-Caraballo, I.; Wróbel, J.S.; Dudarev, S.L.; Nguyen-Manh, D.; Rivera-Díaz-del-Castillo, P.E.J. Interatomic spacing distribution in multicomponent alloys. Acta Mater. 2015, 97, 156–169. [Google Scholar] [CrossRef]
- Widom, M. Prediction of structure and phase transformations. In High-Entropy Alloys; Springer: Cham, Switzerland, 2016; pp. 267–298. [Google Scholar]
- Song, H.; Tian, F.; Wang, D. Thermodynamic properties of refractory high entropy alloys. J. Alloy. Compd. 2016, 682, 773–777. [Google Scholar] [CrossRef]
- Dutta, B.; Ghosh, S. Phonon spectra of PdxFe1−x alloys with transferable force constants. J. Phys. Condens. Matter 2009, 21, 395401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zacherl, C.L.; Shang, S.; Chen, L.Q.; Liu, Z.K. Phonon dispersions in random alloys: A method based on special quasi-random structure force constants. J. Phys. Condens. Matter 2011, 23, 485403. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Phase | Method | V0 (A3) | B0 (GPa) | B′ | ΔErelax (meV) | ΘD (K) |
|---|---|---|---|---|---|---|
| B2(MoW;NbTa) | PAW + SQS | 16.98 | 233 | 4 | - | 296 |
| PAW + SQS + relax | 16.97 | 233 | 4 | 0.6 | 296 | |
| A2 solid solution | PAW + SQS | 16.97 | 232 | 4 | - | 296 |
| PAW + SQS + relax | 16.99 | 231 | 4 | 12.5 | 295 | |
| PAW + SQS + relax [30] | 17.08 | 228 | n/a | n/a | n/a | |
| Experiment [4] | 16.70 | 220 | - | - | - |
| Method | (meV) | (K) | (K) | |
|---|---|---|---|---|
| PAW + SQS | 42.8 | 717 | 717 | 717 |
| PAW + SQS + relax | 30.9 | 517 | 508 | 508 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Körmann, F.; Sluiter, M.H.F. Interplay between Lattice Distortions, Vibrations and Phase Stability in NbMoTaW High Entropy Alloys. Entropy 2016, 18, 403. https://doi.org/10.3390/e18080403
Körmann F, Sluiter MHF. Interplay between Lattice Distortions, Vibrations and Phase Stability in NbMoTaW High Entropy Alloys. Entropy. 2016; 18(8):403. https://doi.org/10.3390/e18080403
Chicago/Turabian StyleKörmann, Fritz, and Marcel H.F. Sluiter. 2016. "Interplay between Lattice Distortions, Vibrations and Phase Stability in NbMoTaW High Entropy Alloys" Entropy 18, no. 8: 403. https://doi.org/10.3390/e18080403