Optimization of Protein Therapies by Polymer-Conjugation as an Effective DDS

1
Graduate School of Pharmaceutical Sciences, Osaka University. 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
2
National Institute of Health Sciences, Osaka Branch Fundamental Research Laboratories for Development of Medicine. 7-6-8 Saito-Asagi, Ibaraki, Osaka 567-0085 Japan
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(1), 162-180; https://doi.org/10.3390/10010162
Submission received: 30 June 2004
/
Revised: 27 September 2004
/
Accepted: 12 December 2004
/
Published: 31 January 2005
(This article belongs to the Special Issue Macromolecules Applied to Pharmaceutical Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Due to recent advances in disease proteomics, many disease-related proteins have been found. It is expected that there will be therapeutically useful proteins among them. However, it is clinically difficult to use most proteins as effective and safe drugs because of their very low stability and pleiotropic actions in vivo. To promote disease proteomic based drug development for protein therapies, we have attempted to develop an optimal polymer-conjugation system for improving the therapeutic potency of proteins. In this review, we introduce this innovative protein-drug system.
Introduction
With the success of the human genome project, the focus of life science research has shifted to the functional and structural analyses of proteins, such as disease proteomics. Therapeutic application of bioactive proteins, such as newly identified proteins and cytokines, are also promising [1,2,3,4,5,6,7]. However, because these proteins are generally quite unstable in vivo, their clinical application requires frequent administration at high dosages. This administration markedly destroys homeostasis, resulting in unexpected side effects. In addition, since bioactive proteins exhibit diverse pharmacological actions in various tissues, it is difficult to obtain selectively only the favorable in vivo actions. For these reasons, clinical applications of bioactive proteins have been limited [7,8,9,10,11].
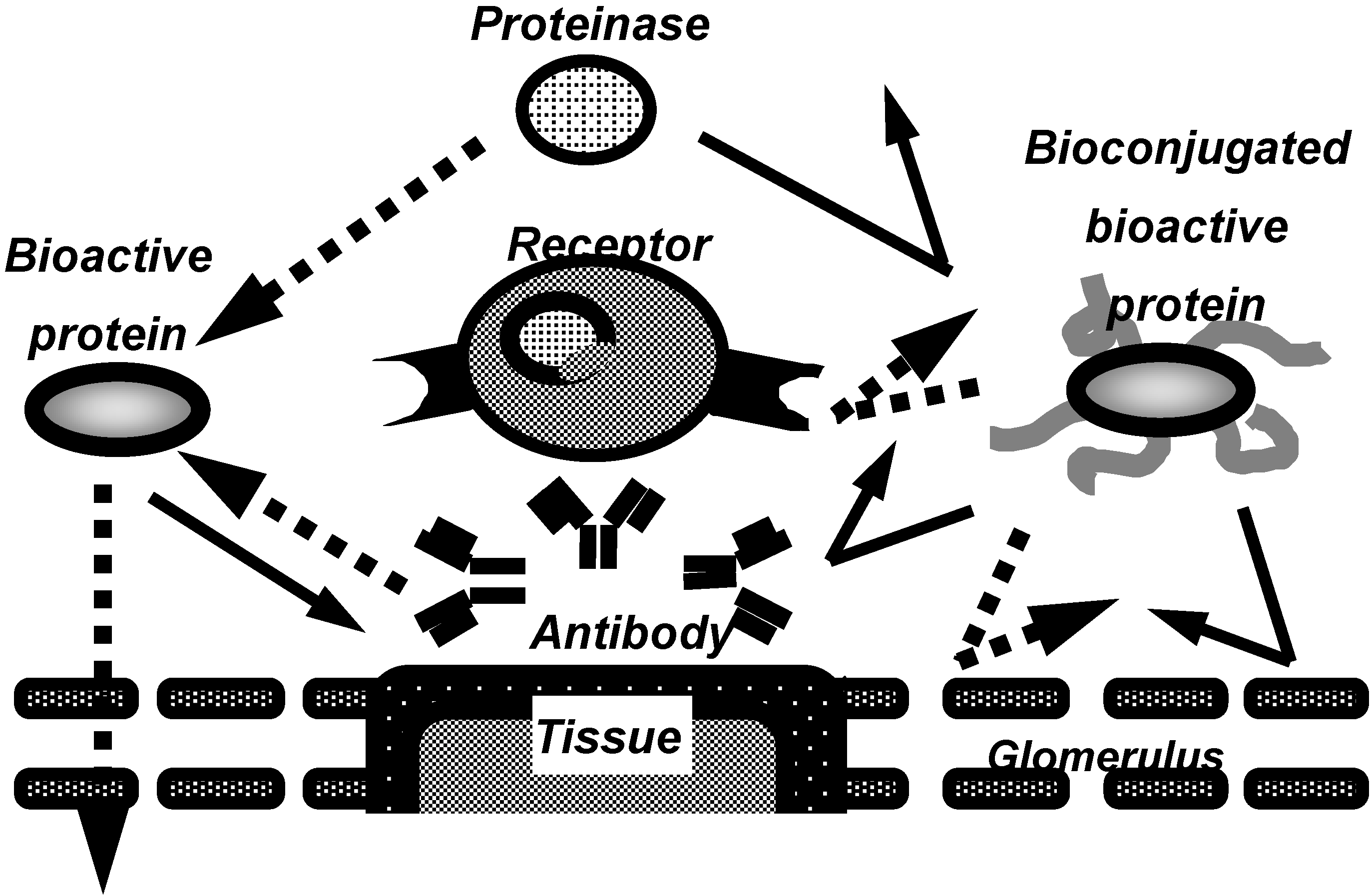
Figure 1.
Characteristics of bioconjugated proteins.
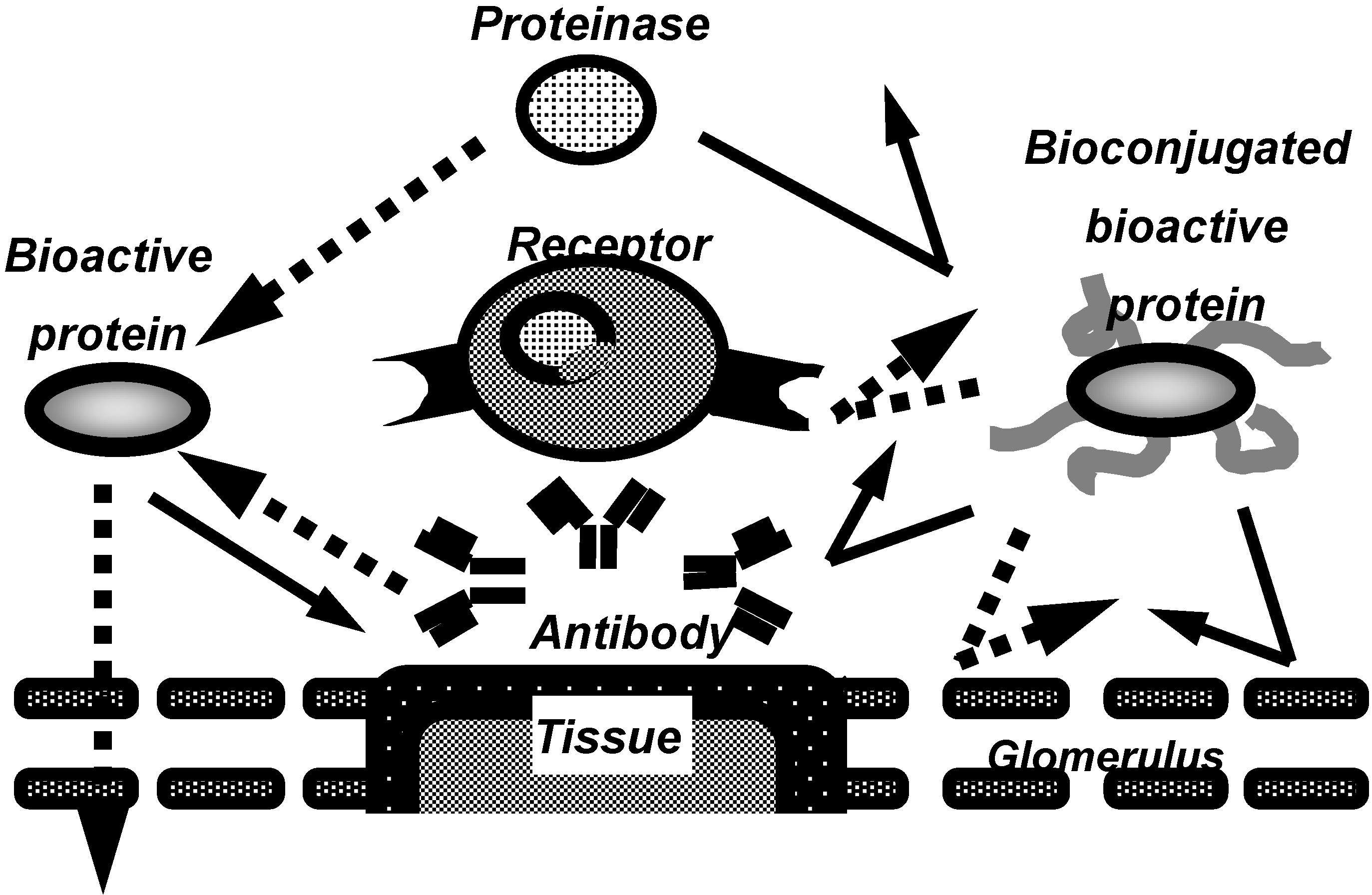
In recent years, to overcome these problems, the conjugation of proteins with water soluble polymeric modifiers has been developed (Figure 1), especially, the conjugation with polyethylene glycol (PEG), often called “PEGylation”. Bioconjugation of proteins with water-soluble polymeric modifiers increases their molecular size and steric hindrance, both of which are dependent on the polymeric modifiers attached the proteins. These effects improve the plasma half-lives of proteins and their stability against proteolytic cleavage, and also decrease their immunogenicity. This allows the therapeutic dose and frequency to be decreased.
In fact, PEGylated interleukin-2, PEGylated interferon and PEGylated adenosine deaminase have demonstrated markedly improved therapeutic efficacy over the native forms, and clinical applications have already been realized [12,13,14,15,16]. However, clinical use of polymer-conjugated proteins has been limited yet. This is due to the conflicting effects of polymer-conjugation of bioactive proteins; conjugation with a polymeric modifier inhibits the transport from blood to tissues and the binding to their receptors. In addition, specific activities of proteins are decreased by the attachment of polymeric modifiers to active sites. Therefore, determination of the relationships among the degree of modification, molecular size, and specific activity is very important to optimize the modification-condition, which enable designing of polymer-conjugated proteins applicable to clinical use.
On the other hand, for further enhancement of the therapeutic potency and safety of polymer-conjugated proteins, more precise control of the in vivo behavior of each protein is necessary for selective expression of their therapeutic bioactivities. We found that polymer-conjugated proteins can be greatly affected by the properties of the polymeric modifiers attached to the surface of the proteins. Therefore, it is necessary to identify appropriate polymeric modifiers for design of conjugated proteins with desirable in vivo behavioral characters. PEG is a low toxicity and low antigenicity polymeric modifier that has been used frequently for conjugation of proteins. From the viewpoint of a drug delivery system, PEG, however, also has some disadvantages as a drug carrier, principally the fact that PEG only has a functional group at the end of the chain, limiting the possibilities of adding new functions to the drugs to control more precisely their pharmacokinetics and tissue distribution. Therefore, alternative water-soluble polymeric modifiers in which new functions, such as targeting and release control of drugs, can be added are required for further development of polymer-conjugated drugs.
In this review, we, at first, show the fundamental information enabling us to design the conjugated bioactive proteins applicable to therapeutic use, taking tumor necrosis factor-alpha (TNF-alpha) and interleukin-6 (IL-6) as examples [17,18,19], and a novel method using the reversible amino-protective reagent dimethylmaleic anhydride (DMMAn) to prevent the decrease of bioactivity by attachment of polymeric modifiers [20]. Next, we show the usefulness of PVP in achieving long plasma half-lives and for application to the tissue-targeting polymer [21,22,23].
PEGylation of bioactive proteins
a) PEG-TNF-alpha
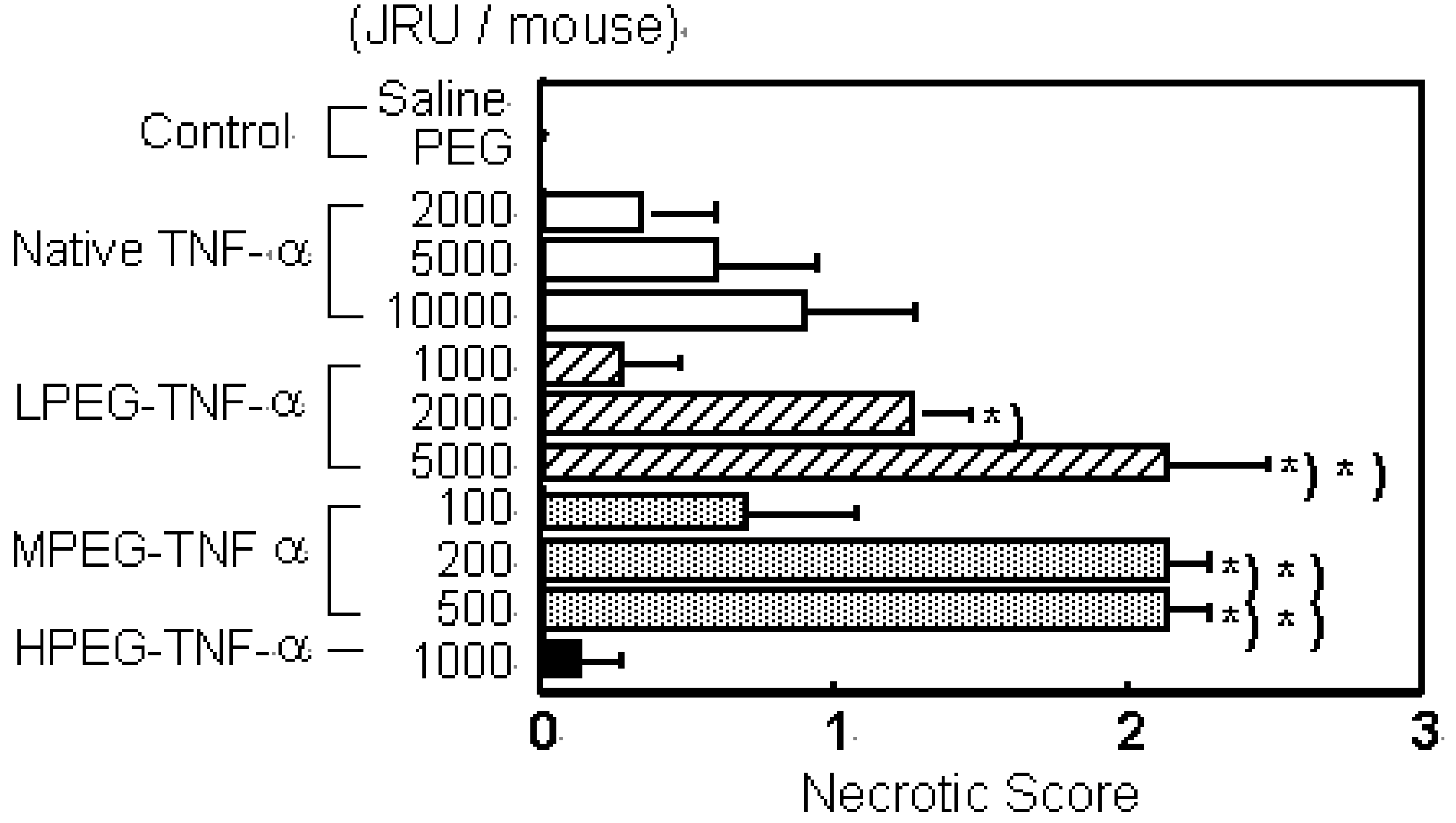
TNF-alpha is a cytokine that was discovered to specifically injure tumors and was thus highlighted as a novel anti-tumor agent [24,25]. However, due to its very low in vivo stability, the continuous-infusion or frequent administration at high doses of TNF-alpha was needed [26,27]. Intravenous TNF-alpha administration has caused marked side effects such as fever, nausea, vomiting, a decrease in blood pressure, and endotoxin-like shock [10,28]. The anti-tumor effects of TNF-alpha result not only from its direct cytotoxic action against various tumor cells, but also from activation of anti-tumor effector immune cells in the blood and specific damage to the tumor vessels. In addition, in the process of bleeding necrosis in the tumor vessels, the vascular permeability of the tumor vessels is selectively increased, promoting transport from blood to the tumor tissue. Therefore, improvement in blood stasis may enhance all anti-tumor action mechanisms of TNF-alpha increasing its bioavailablity. Therefore, we performed conjugation of the lysine amino residues of TNF-alpha using PEG [18]. In PEGylation of TNF-alpha, the specific activity of PEGylated TNF-alpha decreased with the PEG modification rate. Additionally, when the PEG modification rates (degree of PEG-modification) are the same, the bioactivity of PEGylated TNF-alpha decreased with an increase in the molecular size of the attached PEG [17]. Our other studies on PEGylated IL-6 and leukemia inhibitory factor (LIF) yielded similar results [18]. Thus, in bioactive proteins such as TNF-alpha, IL-6 and LIF that require binding to a receptor for the expression of activity, consideration should be given to inhibition of activity derived from inhibition of binding to receptor molecules caused by steric hindrance by the polymeric modifier, in addition to a decrease in the specific activity due to modification of the lysine residues. On the other hand, the in vivo anti-tumor effects of PEGylated TNF-alpha was the most marked for MPEG- TNF-alpha (molecular weight; Mn=108,000) obtained by PEGylation using PEG with a molecular weight of 5,000 (PEG5000) (Figure 2a).
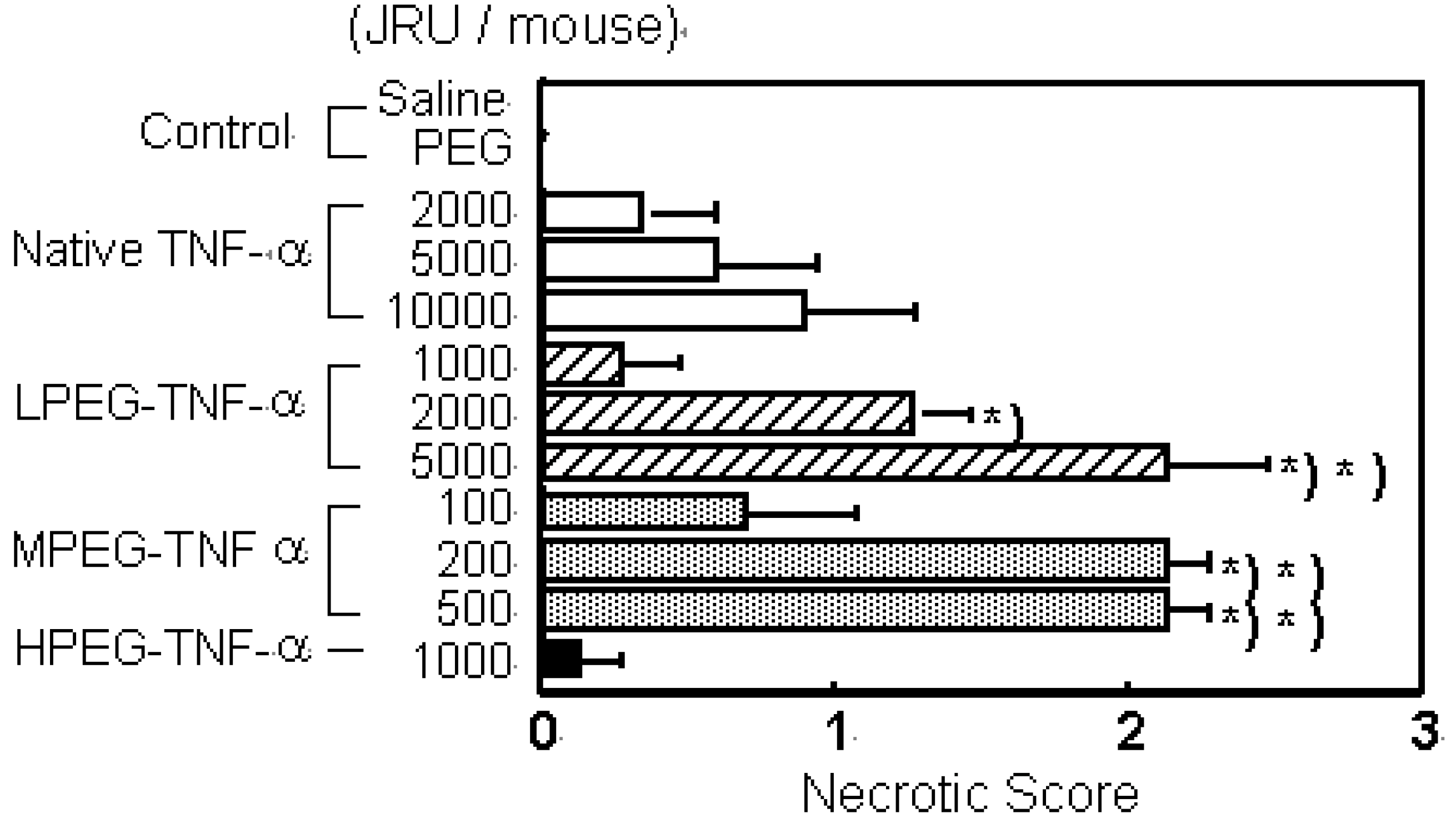
Figure 2.
PEGylation of TNF-α and IL-6 selectively increase their therapeutic effects. a) Tumor necrosis effects of native TNF-α and PEG-TNF-αs on Meth-A solid tumors. Each value represents mean ± S.E. ∗P<0.001, ∗∗P<0.05 significantly different from the group treated with 10,000 JRU of native TNF-α.
Figure 2.
PEGylation of TNF-α and IL-6 selectively increase their therapeutic effects. a) Tumor necrosis effects of native TNF-α and PEG-TNF-αs on Meth-A solid tumors. Each value represents mean ± S.E. ∗P<0.001, ∗∗P<0.05 significantly different from the group treated with 10,000 JRU of native TNF-α.
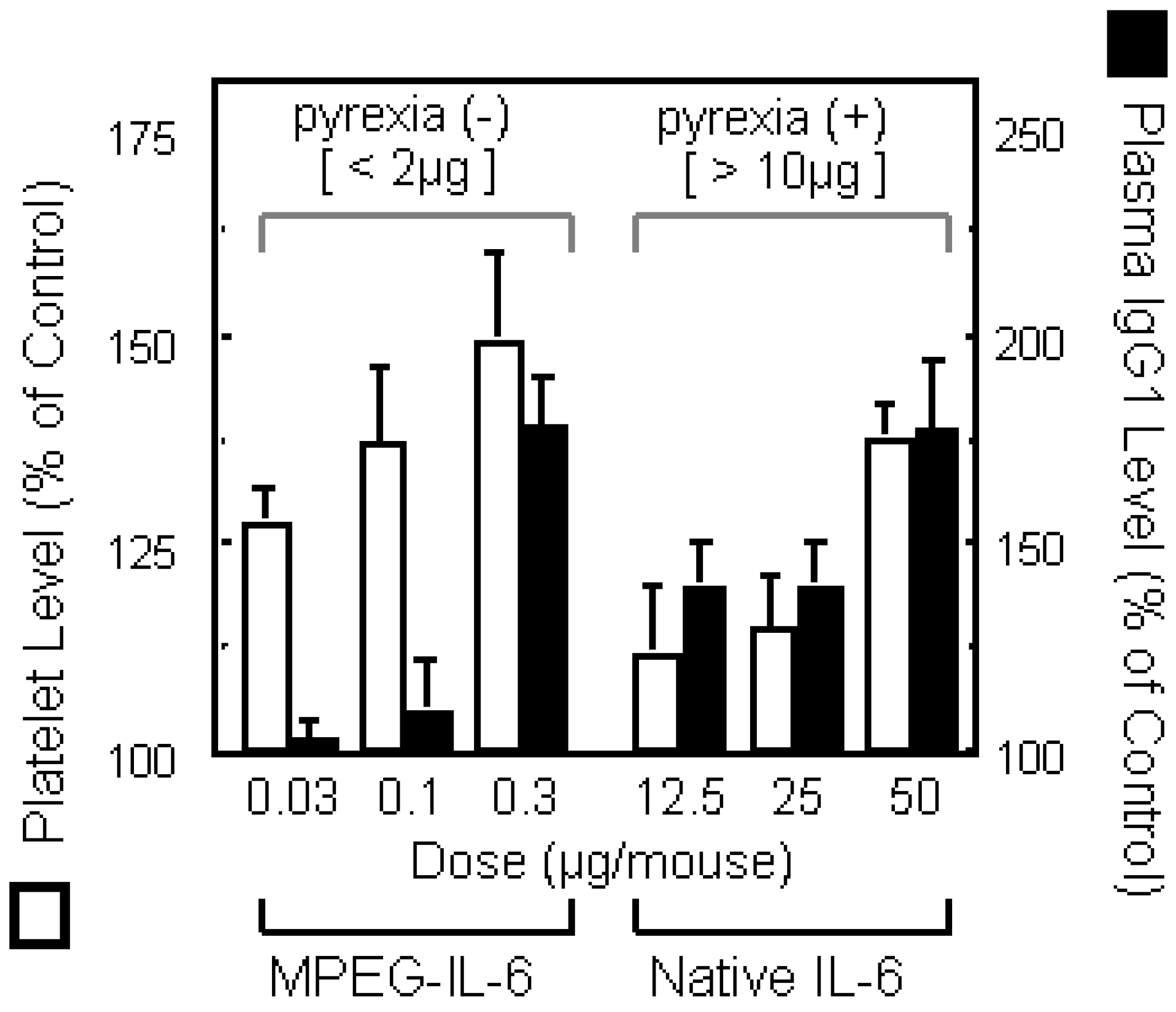
b) Effect of PEG5000-IL-6 and Native IL-6 on platelet production (favorable effect) or plasma IgG1 depressions (side effect) were compared. On day 9, blood was taken form tail vein of mice injected of native IL-6 or PEG5000-IL-6 Fr.4 every 2 days for a week.
b) Effect of PEG5000-IL-6 and Native IL-6 on platelet production (favorable effect) or plasma IgG1 depressions (side effect) were compared. On day 9, blood was taken form tail vein of mice injected of native IL-6 or PEG5000-IL-6 Fr.4 every 2 days for a week.

Its anti-tumor activity was 100 times higher than that of the unmodified TNF-alpha without increasing the toxic side effects. No marked enhancement of anti-tumor effects were observed for slightly modified LPEG-TNF-alpha (Mn=84,000) with a high specific activity or excessively modified HPEG-TNF-alpha (Mn=122,000) with a high molecular size, though they were also obtained after conjugation of PEG with a molecular weight of 5,000. Conjugation using PEG with a molecular weight of 2,000 or 12,000 did not produce PEGylated TNF-alpha s comparable to MPEG-TNF-alpha. These results suggest that PEGylation of TNF-alpha does not always produce marked in vivo anti-tumor effects, but there is an optimal molecular weight of the polymeric modifier and an optimal modification rate - molecular size - activity correlation of PEGylated TNF-alpha.
b) PEG-IL-6
When IL-6 is utilized therapeutically to promote the formation of platelets, its targets are the megakaryocytes [29,30,31]. It is known that megakaryocytes express large amounts of high affinity IL-6 receptors (IL-6 receptor-gp130 complex) [32,33], and that they are abundant on the pulmonary vascular lumens and on the outer surfaces of marrow veins. Therefore, if IL-6 is modified to remain longer in the blood and thus smaller dose levels are required for therapeutic use, it will be possible to make the in vivo distribution and receptor affinity of IL-6 such that a selective and efficient action of IL-6 on megakaryocytes can be achieved. Furthermore, since the transfer of IL-6 into the liver and spleen cause adverse reactions [34,35,36], the improved retention of IL-6 in the blood and the resultant decrease in the transfer and accumulation of IL-6 to these tissues is expected to reduce the side effects of IL-6 therapy. Thus, we attempted the conjugation of IL-6 with PEG, to increase in the activity of IL-6 in the promotion of platelet production and to reduce its side effects. When IL-6 was subjected to PEGylation under optimum conditions, selected by consideration of the relationships between specific activity, degree of PEG-modification, molecular size, etc., the resultant PEG-modified IL-6 (MPEG-IL-6) showed plasma half-life more than 100 times greater than that of native IL-6. MPEG-IL-6 showed more than 500 times the thrombopoietic potency of native IL-6 (Figure 2b). Furthermore, strong adverse reactions such as fever, IgG production and acute protein production observed following administration of native IL-6, were seldom seen after administration of MPEG-IL-6. These results suggested separation of therapeutically favorable targeted actions from side effects by means of PEGylation has been successful. It was found that PEGylation allows exertion of selected favorable actions of cytokines via the following mechanisms: (1) improved in vivo stability reduces the dose level and thus the blood level of cytokines, making it possible for a given cytokine to exert its selected actions on the basis of differences in the affinity of the cytokine for various receptors; and (2), regulation of the behavioral characteristics (blood retention and tissue transfer) of a given cytokine enables the cytokine to exert selected actions depending on differences between its distribution in different tissues. We have thus succeeded in making cytokines useful as therapeutic agents by improving their stability in vivo and increasing in selected favorable actions (anti-tumor activity in the case of TNF-alpha and the promotion of platelet production in the case of IL-6). These results suggested that polymer-conjugation is a pragmatic approach to successful therapies with various bioactive proteins and peptides.
A novel polymer-conjugation technique
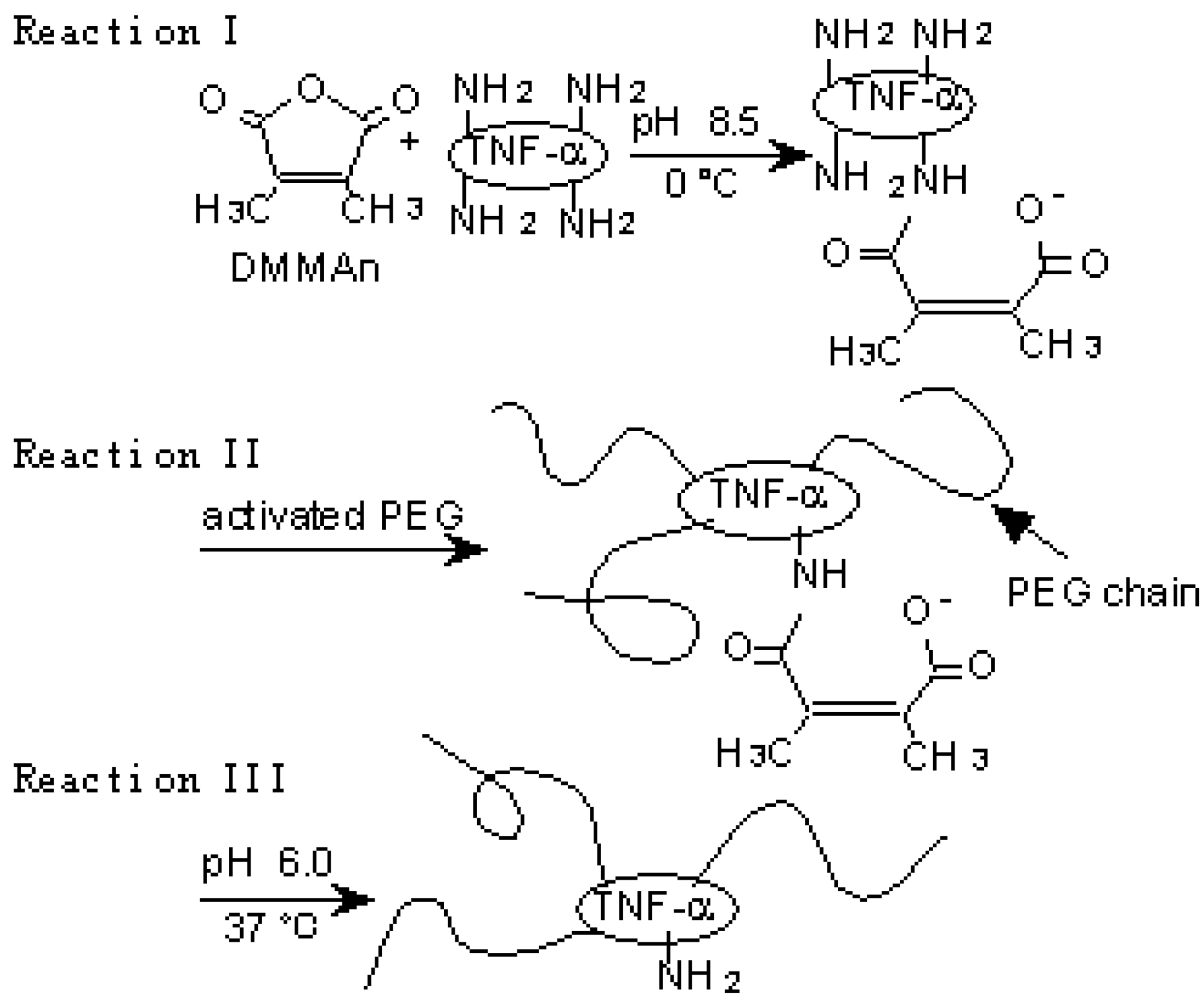
Lysine amino groups of proteins are often used as binding sites of PEG because they are highly reactive and the PEGylation reaction is mild enough to minimize disruption of the protein structure [37]. This PEGylation, however, is nonspecific and occurs at the N-terminus as well as internal lysine residues, some of which may be in or near protein active sites, resulting in loss of specific bioactivity. Therefore, conjugation methods which avoid modification of amino groups around the receptor binding region of proteins are needed. We attempted to control modification by using the reversible amino-protective reagent dimethylmaleic anhydride (DMMAn) [20]. PEG-TNF-alpha(+) was prepared according to the reactions presented in Figure 3a.
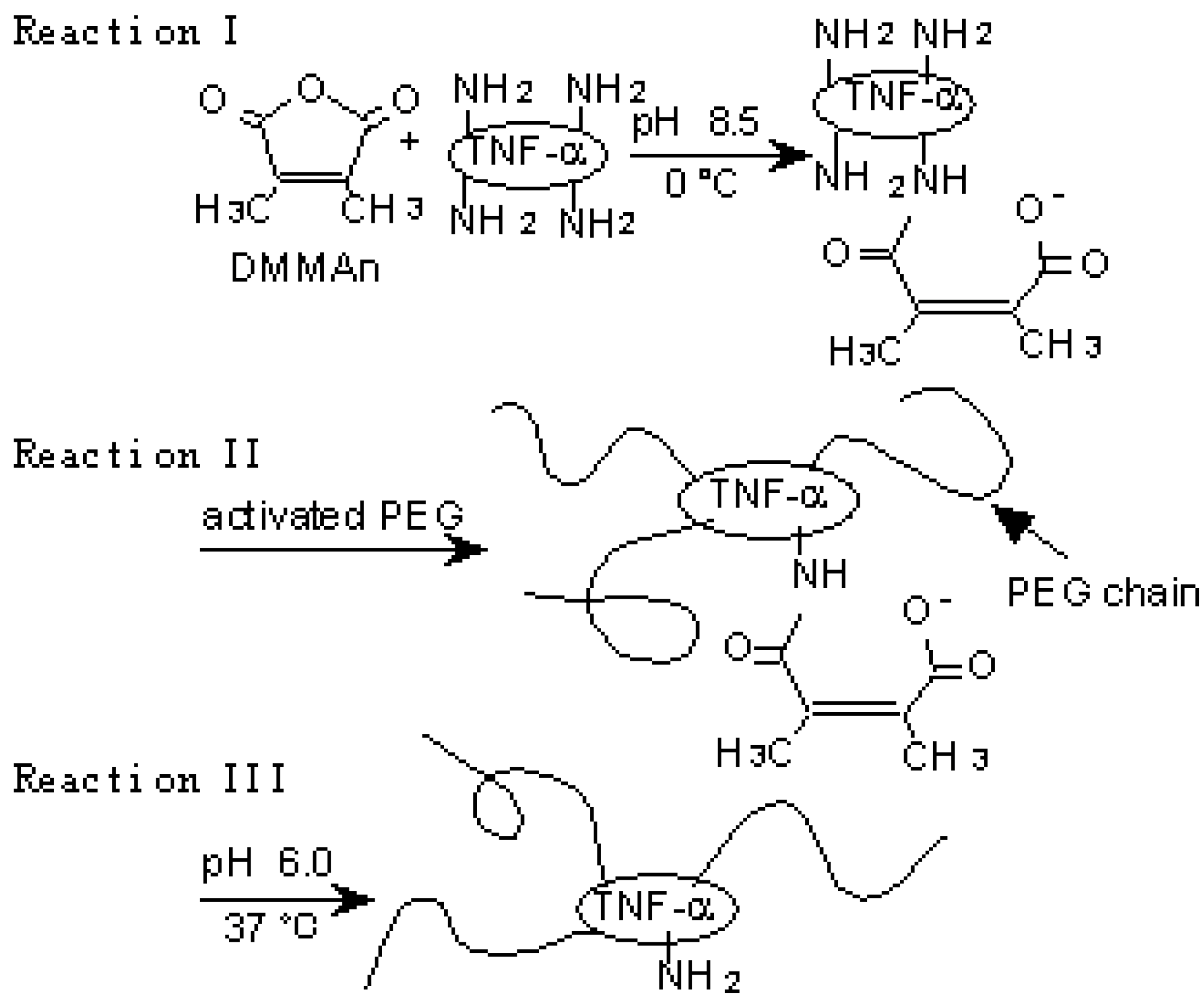
Figure 3.
A novel polymer-conjugation technique with a reversible amino-protective reagent. a) Schematic protocol of PEGylation of TNF-α using DMMAn: Reaction I, protection of partial amino groups by DMMAn; Reaction II, PEGylation to remaining lysine amino groups; Reaction III, regeneration of amino groups by releasing DMMAn.
Figure 3.
A novel polymer-conjugation technique with a reversible amino-protective reagent. a) Schematic protocol of PEGylation of TNF-α using DMMAn: Reaction I, protection of partial amino groups by DMMAn; Reaction II, PEGylation to remaining lysine amino groups; Reaction III, regeneration of amino groups by releasing DMMAn.
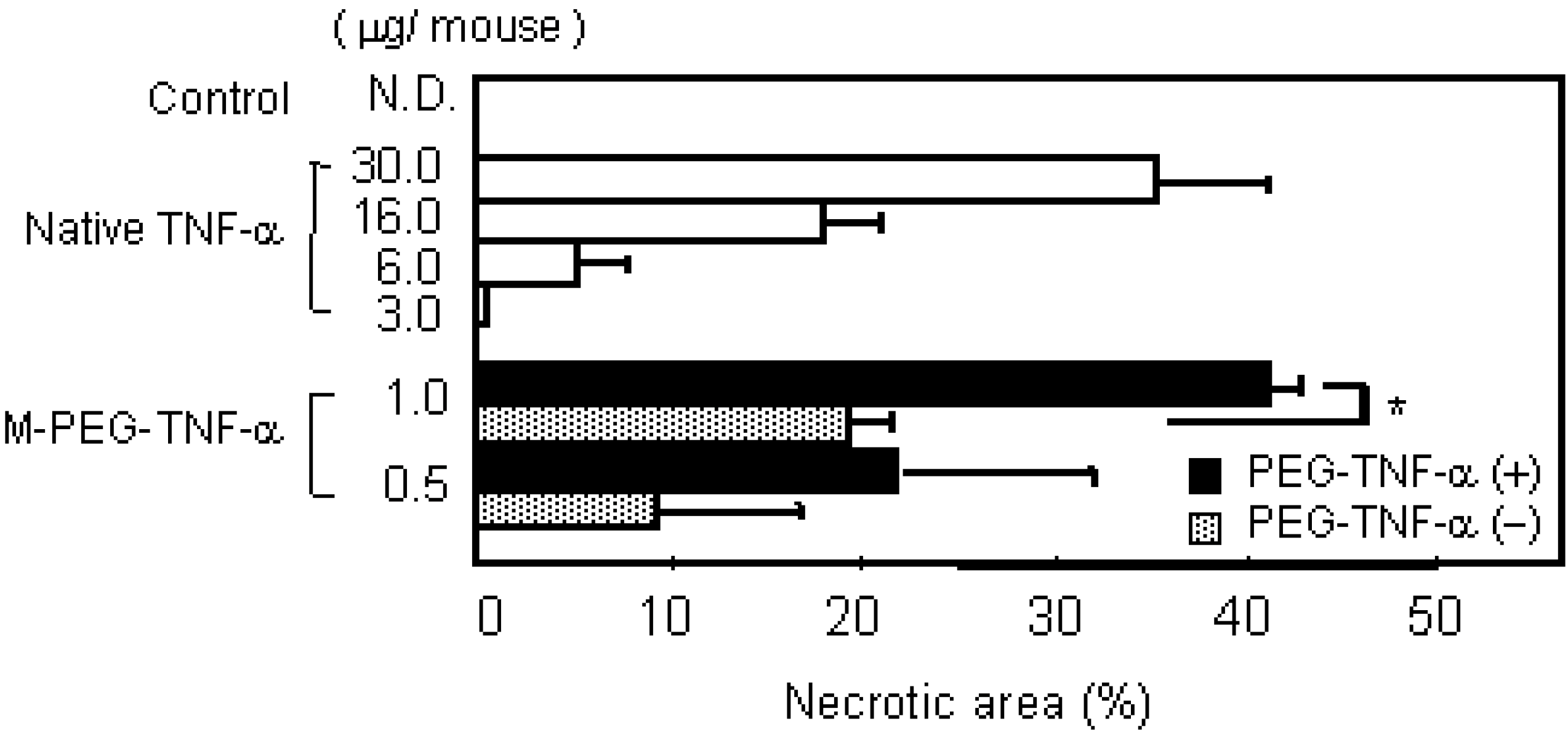
b) Tumor necrotic effects of PEG-TNF-αs on Meth-A solid tumor in mice. Meth-A-bearing BALB/c mice were given native TNF-α, PEG-TNF-α(-),or PEG-TNF-α(+) i.v. The control group was given saline. The area of hemorrhagic necrosis was measured 24h after injection. Each value is the mean ± S.E. of four animals. ∗P < 0.01, statistical significance compared with PEG-TNF-α(-)s.
b) Tumor necrotic effects of PEG-TNF-αs on Meth-A solid tumor in mice. Meth-A-bearing BALB/c mice were given native TNF-α, PEG-TNF-α(-),or PEG-TNF-α(+) i.v. The control group was given saline. The area of hemorrhagic necrosis was measured 24h after injection. Each value is the mean ± S.E. of four animals. ∗P < 0.01, statistical significance compared with PEG-TNF-α(-)s.

The protection of partial amino groups in TNF-alpha by DMMAn was confirmed by fluorimetric analysis. Then, to examine whether the active core of TNF-alpha can be protected from PEGylation using DMMAn, changes in specific activity were examined in vitro. Specific activities improved for all fractions of PEG-TNF-alpha(+) compared with the similar molecular size fractions of PEG-TNF-alpha(-) tested. This result indicates that the use of DMMAn improves cytokine receptor binding. We found similar results for PEGylation of IL-6 and granulocyte macrophage colony-stimulating factor. To examine the influence of improvements in specific activity in vivo, Meth-A fibrosarcoma bearing mice were given native and PEG-TNF-alphas (Figure 3b). The antitumor effects of MPEG-TNF-alpha(+) were 2-fold more potent than those of MPEG-TNF-alpha(-), which was the most potent TNF-alpha not treated with DMMAn, and 30-fold more potent than those of native TNF-alpha. Significantly, improvements in the specific activity of MPEG-TNF-alphas were only about 50%, but improvements in antitumor effects were more than 2-fold in vivo. These results suggested that our method is easy and useful for the clinical application of polymer-conjugated cytokines.
Screening of polymeric drug carriers
It is well known that the fate and distribution of the conjugates can be attributed to the physicochemical properties of polymeric modifiers such as molecular weight, electric charge, and hydrophilic-lipophilic balance [38]. The increase of therapeutic effects of drugs conjugated with polymeric modifiers is attributed to the pharmacokinetics of conjugated drug. Therefore, selecting the polymeric modifier by considering the influence of physicochemical characteristics on the pharmacokinetics of the polymeric modifier is markedly important. As mentioned above, sequential and multiple strategies are needed for optimization of drug therapy based on polymer-conjugation: i) optimum selection of polymeric modifier considering the disposition of drugs and objective such as targeting or controlled release, ii) polymer-conjugation based on estimation of characterization such as molecular size, modification site, degree of modification, and specific activity, and iii), assessment of therapeutic effect and pharmacokinetics of conjugated drug.
As mentioned, PEG is a low toxicity and low antigenicity polymeric modifier that has been frequently used for polymer-conjugation. PEG is a polyether diol of general structure HO-(CH2CH2O)n-H, where functionalization of PEG is restricted to the utilization of the terminal primary OH groups [39]. From this viewpoint, modifiable polymeric modifiers are needed to control the biopharmaceutical characteristics of conjugated drugs. Therefore, we assessed the pharmacokinetic profile of various water-soluble polymers with molecular sizes of about 5,000, and compared their pharmacokinetics to PEG5000 [40].
First, the elimination profile of 125I-labeled various polymers with the same molecular size after i.v. injection in mice bearing S-180 solid tumors was studied. The polymer formulations used to evaluate these are: PEG, polyvinylpyrrolidone (PVP), polyacrylamide (PAAm), polydimethylacrylamide (PDAAm), polyvinyl alcohol (PVA), and dextran. PVP, PAAm, and PDAAm are functionalized by introduction of various comonomers on radical polymerization. PVA and dextran have many primary OH groups that can be used for conjugation on the side chain.
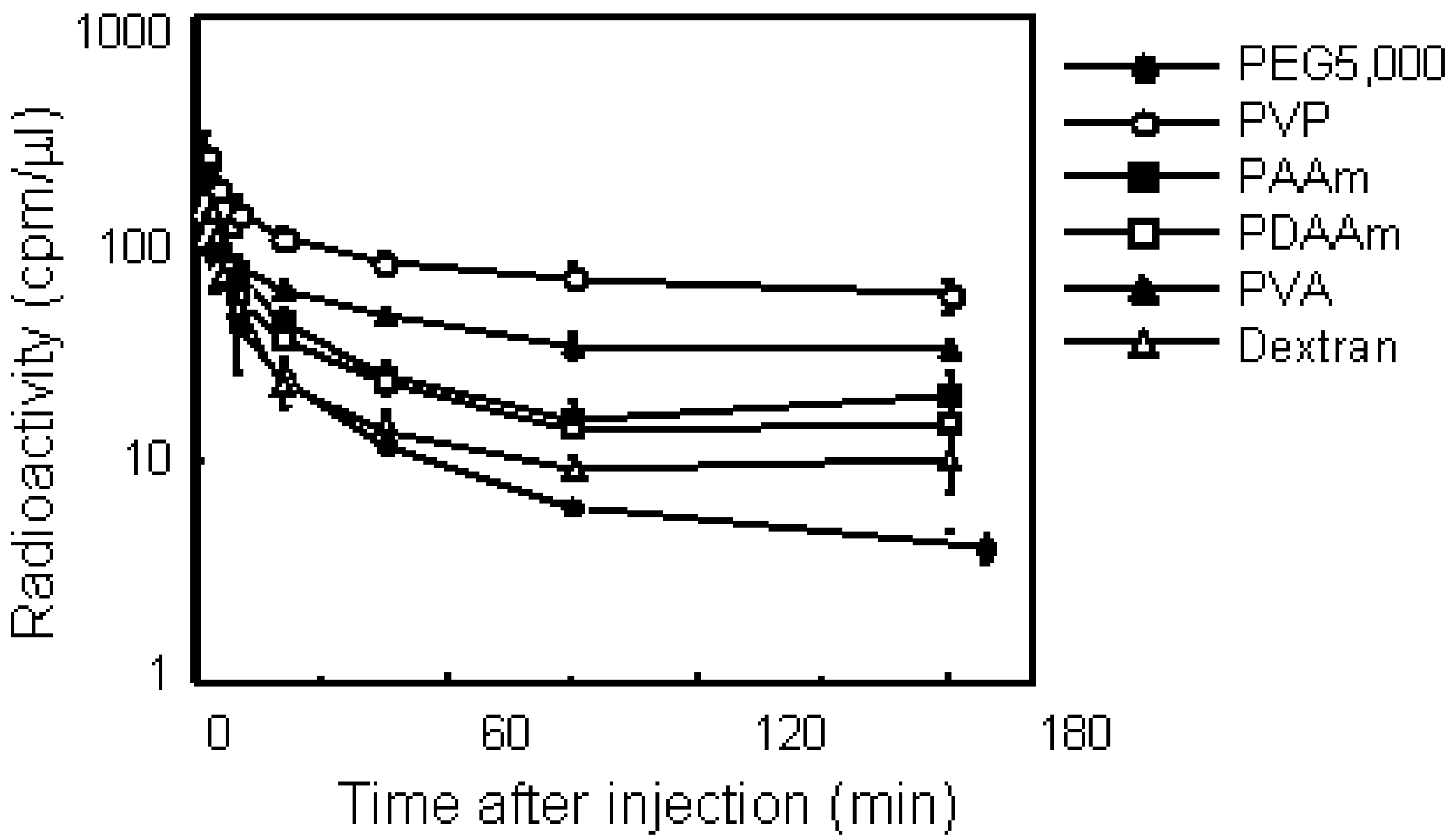
Figure 4.
Plasma clearance of various water-soluble polymers in mice bearing S-180 solid tumors after i.v. injection. Mice were intravenously injected with 125I-labeled polymer. After administration, blood was collected from the tail vein at indicated times and the radioactivity was measured by a γ-counter. Mice were used in groups of five. Each value is mean ± SD.
Figure 4.
Plasma clearance of various water-soluble polymers in mice bearing S-180 solid tumors after i.v. injection. Mice were intravenously injected with 125I-labeled polymer. After administration, blood was collected from the tail vein at indicated times and the radioactivity was measured by a γ-counter. Mice were used in groups of five. Each value is mean ± SD.
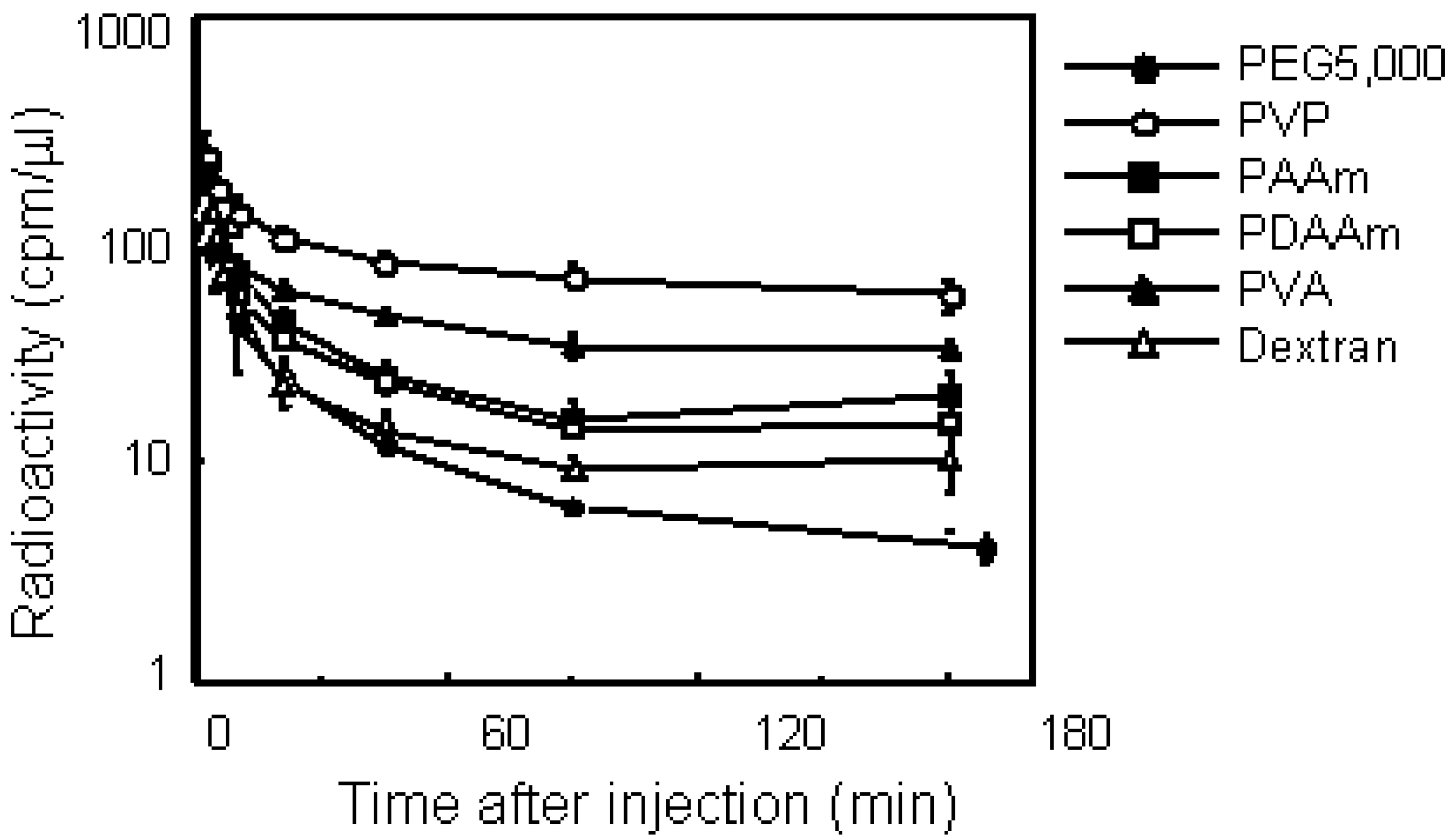
Figure 4 illustrates the plasma clearance of various polymers. All polymers showed biphasic elimination patterns. PEG5000 and dextran, which are used frequently as drug carriers, were eliminated rapidly from the blood circulation. On the other hand, PVA and PVP circulated longer than the other polymers, while these nonionic polymers had the same molecular size of PEG5000. PVP exhibited the longest residence of all the polymers in this study, and 25% of the injected dose remained after 180 min. Pharmacokinetic analysis revealed definite differences among each polymer with respect to plasma clearance and tissue distribution. PVP showed the longest MRT of all polymers examined. The total clearance of PVP was about 10-fold lower than that of PEG5000. The distribution volume of dextran was the highest of all these polymers; its volume was double that of PVP. We next studied the tissue distribution of polymers 3 h after i.v. injection. Although all polymers with the same molecular weight dispercity in this study were nonionic and water-soluble, each polymer showed a characteristic distribution. PEG and PVP did not exhibit specific tissue accumulation, on the other hand, dextran was accumulated in the liver and spleen. PVA and PAAm also had no specific distribution, but PDAAm tended to accumulate in the kidney. As demonstrated clearly, dextran is not appropriate for prolonging the circulation time of drugs.
PVP has the longest circulation time and, its tissue distribution was extremely restricted. In addition, it is easy to introduce various comonomers on radical polymerization to PVP. These results suggest that PVP is the most feasible polymeric modifier for localizing the conjugated drug in blood.
Conjugation with PVP
a) PVP- TNF-alpha
To design conjugated proteins with greater efficacy and safety, i.e. to selectively enhance desirable therapeutic activities of bioactive proteins without increasing their side effects, it is important to closely regulate their in vivo behavioral characteristics, taking into account their mechanism of action. Thus, the development of polymeric modifiers with useful DDS (Drug Delivery System)-functions, which can regulate their in vivo behavioral characteristics such as targeting capability and controlled release, is required. Therefore, we examined other candidates as polymeric modifiers for introduction of these new functions. As mentioned above, polyvinyl pyrrolidone (PVP), to which useful functional groups can be introduced by radical co-polymerization, was found to be retained in blood better than PEG.. Furthermore, PVP is also highly biocompatible, and is widely used as a plasma expander. With this in mind and to assess the usefulness of PVP as a polymeric modifier for protein conjugation, we evaluated the therapeutic potency of PVP-TNF-α compared with that of PEG conjugate (PEG-TNF-α) [21,22]. The terminal carboxyl-bearing PVP was prepared by radical polymerization of VP. The number-average molecular weight (Mn) of the synthesized PVP was controlled by varying the amount of transfer agent added to the reaction. Previously, we showed that PEG- TNF-alpha and PEG-IL-6, conjugated with PEG (5,000 Mn), possesed marked antitumor activity or thrombopoietic activity, respectively. These conjugates were revealed to have higher therapeutic effects in vivo than those prepared with PEG (2,000 Mn) or PEG (12,000 Mn). Therefore, in this study, 6,000 Mn PVP purified by gel filtration was used as a polymeric modifier for the first choice.
Natural human TNF-alpha was conjugated with activated PVP (Mn; 6,000, Mw/Mn =1.14) via amide bonds between lysine amino groups of TNF-alpha and N-hydroxyscuccinimide groups of PVP at the end of the main chain. The resulting PVP- TNF-alpha was purified from native TNF-alpha and separated into five fractions of various molecular sizes by GF-HPLC (protein standard). The activities of PVP- TNF-alphas decreased with increasing of molecular weight and degree of PVP-modification (PVP-attachment to TNF-alpha). This result was also observed when TNF-alpha was conjugated with PEG, and this profile of changes in the bioactivity of PVP-TNF-alphas was similar to that observed with modification of TNF-alpha with PEG (Mn; 5,000, Mw/Mn =1.32).
The anti-tumor effects of PVP-TNF-alphas on S-180 solid tumors were compared with those of native TNF-alpha and MPEG-TNF-alpha by single i.v. injection. The antitumor effects were evaluated by a score of hemorrhagic necrosis 24 hr after sample administration. The antitumor effect of PVP-TNF-alpha fraction 3 (Mn; 101,000), in which 40% of the total lysine amino groups of TNF-alpha were coupled with PVP, was markedly higher than that of native TNF-alpha at a dose of 10,000 JRU/mouse and induced complete regression in two of seven mice(data not shown).
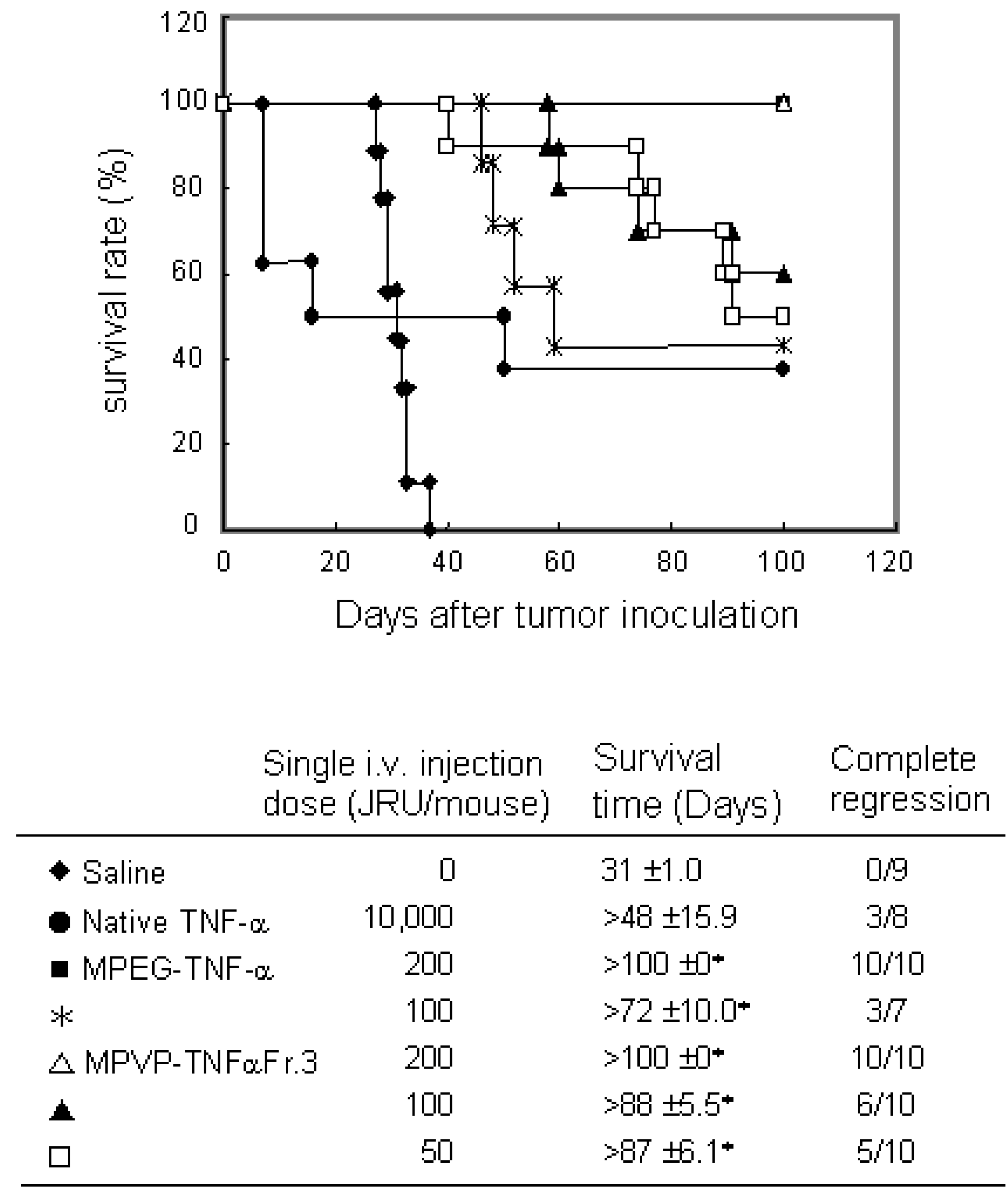
Figure 5.
Antitumor effect of PVP-TNF-α by scheduled administration on survival days after Meth-A tumor inoculation. Meth-A-bearing BALB/c mice were given samples on days 7, 10, 14 and 17 after tumor inoculation. Complete regression was defined when tumor was not regrown for >100 days. Statistical significance compared with saline control: ∗P< 0.01.
Figure 5.
Antitumor effect of PVP-TNF-α by scheduled administration on survival days after Meth-A tumor inoculation. Meth-A-bearing BALB/c mice were given samples on days 7, 10, 14 and 17 after tumor inoculation. Complete regression was defined when tumor was not regrown for >100 days. Statistical significance compared with saline control: ∗P< 0.01.
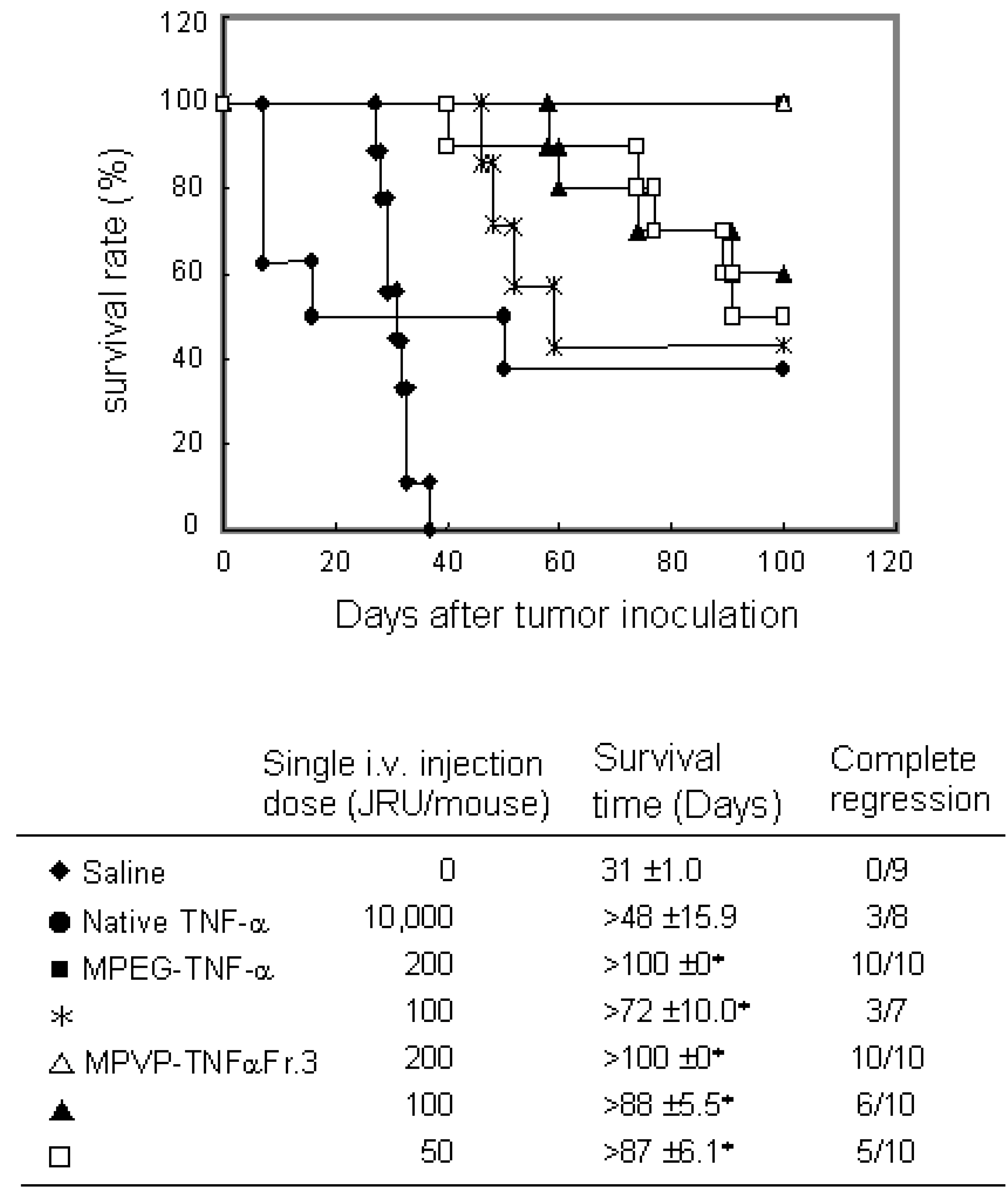
To investigate the usefulness of PVP as a polymeric modifier and PVP-TNF-alpha fraction 3 as a systemic antitumor agent, we compared the antitumor potency of PVP-TNF-alpha fraction 3 to those of native TNF-alpha and MPEG-TNF-alpha with scheduled i.v. injections on Meth-A solid tumors. As shown in Figure 5, PVP-TNF-alpha fraction 3 and MPEG-TNF-alpha at a dose of 200 JRU showed the maximal antitumor effects without any toxic side effects (such as sudden death and others) and had antitumor effects superior to that of native TNF-alpha at a dose of 10,000 JRU. On the other hand, only 50 JRU of PVP-TNF-alpha fraction 3 was needed to exhibit a marked antitumor potency, and tumor growth was completely inhibited for the observation period, as in 10,000 JRU native TNF-alpha and 100 JRU MPEG-TNF-alpha. These results indicated that PVP-TNF-alpha fraction 3 was approximately 200- and 2-fold more potent an antitumor agent than native TNF-alpha and MPEG-TNF-alpha, respectively. The plasma half-life of PVP-TNF-alpha fraction 3 (360 min) was about 80- and 3-fold longer than those of native TNF-alpha (4.6 min) and MPEG-TNF-alpha (122 min), respectively. As described above, anti-tumor effects of TNF-alpha are due not only to direct cytotoxicity against tumor cells, but also to specific injury of the tumor vascular and effective activation of anti-tumor immune cells. TNF-alpha selectively enhances the vascular permeability of tumor vessels. The enhancement of TNF-alpha half-time may lead to a decrease in its distribution to the liver and spleen, which are the major sites of side effects, and would selectively increase its anti-tumor effects. Therefore, we are confident that the increased anti-tumor potency of PVP-TNF-alpha Fr.3 relative to native TNF-alpha and MPEG- TNF-alpha may be attributed to increased half-life. These results suggest that PVP is a useful polymeric modifier for conjugation of TNF-alpha to increase its antitumor potency, and multifunctionally conjugated TNF-alpha may be a potenciated antitumor agent for clinical use.
b) PVP-IL-6
Next, we applied PVP for polymer conjugated-IL-6 and assesed its usefulness as a novel protein modifier [41]. PVP-IL-6 was synthesized and separated into three fractions with different molecular size to assess the appropriate degree of modification for the highest in vivo thrombopoietic potency. To investigate the effects of PVP-modification on therapeutic activity (thrombopoietic activity) of IL-6 in vivo, we administered PVP-IL-6s of various molecular sizes subcutaneously to mice every 2 days for 8 days. Administration of native IL-6 at 50ug/day caused a 30% increase in platelet number with respect to the most potent enhancer of thrombopoieses among the three fractions of PVP-IL-6s examined. No severe side effects such as body weight loss were observed in PVP-IL-6-treated mice. All mice treated with native IL-6 had acute phase reactions and increase in plasma IgG1 level. This result suggested that PVP is efficient polymer for many cytokines.
PVP-derivates
In the DDS area, development of targeting polymers for specific tissues is very important. In order to deliver a conjugated drug to a targeted tissue, the conjugate must display desirable pharmacokinetic characteristics such as plasma clearance and tissue distribution. Therefore, to control the in vivo behavior of conjugated drugs with polymeric modifiers, the pharmacokinetic characteristics of the polymeric modifiers themselves must be assessed. Based on our studies mentioned above, selection of the optimal polymeric modifiers for manifestation of specific characteristics of drugs and for the purpose of drug therapeutic regimens will be obtained.
Polyvinyl pyrrolidone (PVP) can be easily mixed with various co-monomers by radical polymerization, in order to control its physicochemical properties and to add functions such as targeting or sustained release. Additionally PVP is the most suitable polymeric modifier for prolonging the circulation lifetime of a drug and localizing the conjugated drug in blood, as described above. Previously, we found that nonionic polymers did not interact with endothelial cells, but the increase of interaction between polymers and endothelial cells is parallel to the amount of charge or hydrophobic groups. To evaluate the relationship between pharmacokinetics and their anionic functional groups, copolymers of carboxylated PVP [poly(vinylpyrrolidone-co-acrylic acid)] and sulfonated PVP [poly(vinylpyrrolidone-co-vinylsulfonic acid)] were synthesized by radical polymerization [23]. The plasma clearance of PVP and anionized PVPs were compared in mice bearing S-180 solid tumors. The anionized PVP derivatives in the circulation decreased as the number of anionic groups in the PVP increased. Anionized PVPs were cleared more quickly from the blood than PVP, and the clearance of carboxylated PVP from blood was similar to that of sulfonated PVP with the same content of anionic groups. Endothelial cells and the glomerular capillary wall of the kidney are known to be coated with highly polyanionic sialoprotein [42]. Therefore, anionic charged polymers, such as anionized dextran, are generally considered to be cleared more slowly from the circulation than nonionic and cationic polymers despite having the same molecular weight [43]. The reason for this discrepancy is not clear, but it may be partially due to the differences in the structures of anionized polymers. Polysaccharides, such as dextran, have often been used to characterize the pharmacokinetics of polymeric modifiers with various characteristics [44]. Nonionic and cationic polysaccharides are rapidly captured by the cells of the reticuloendothelial system (RES) mainly in the liver after their i.v. injection, but anionized polysaccharides are barely taken up by the RES due to the electrostatic repulsion between the negative charge of the polymer and the vascular wall [13]. Consequently, the plasma half-lives of anionized polysaccharides are longer than those of nonionic and cationic polysaccharides, even if anionized polysaccharides are distributed in the kidney. By contrast, PVP did not show any specific tissue distribution and mainly remained in blood. Thus, the anionized PVPs are suitable for assessing the effects of anionic groups on pharmacokinetic characteristics.
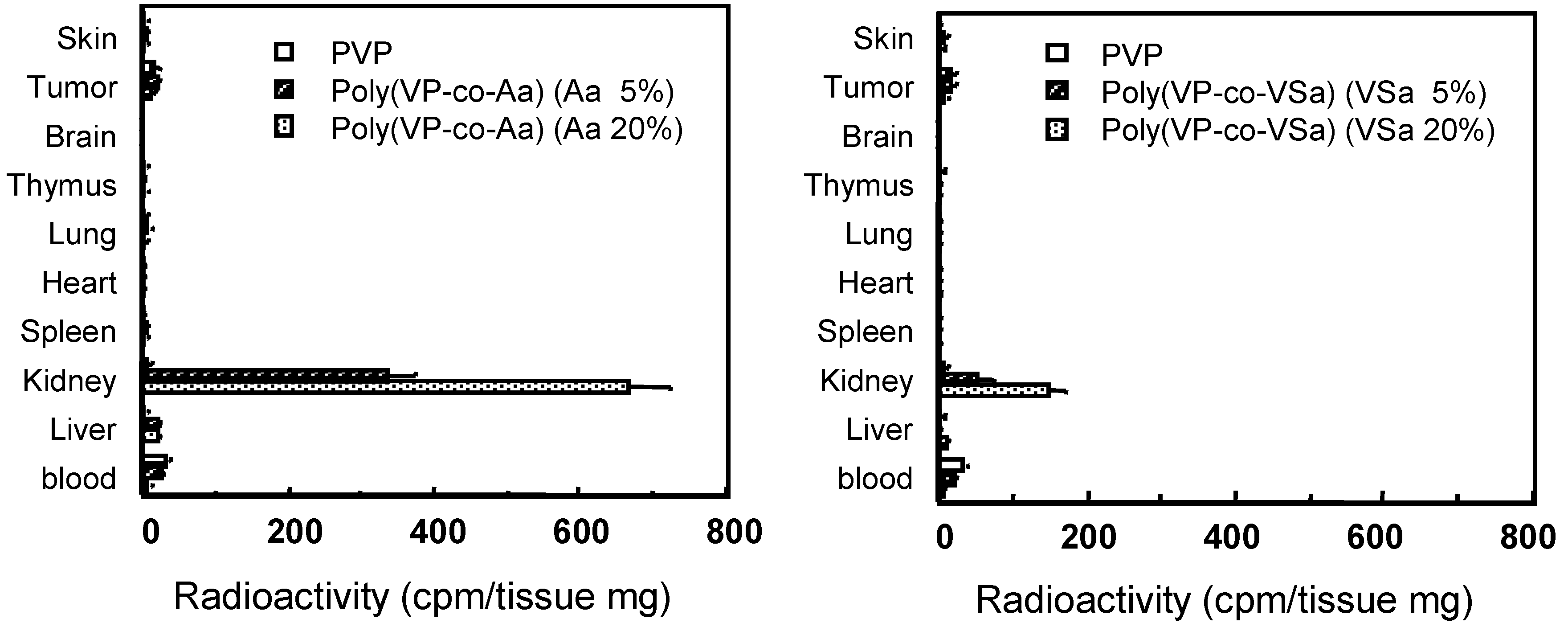
The tissue distribution of PVP and anionized PVPs of the same molecular size were measured 3 h after i.v. injection (Figure 6). Anionized PVPs accumulated more in the kidney than in other tissues, although PVP showed little tissue-specific localization. In particular, carboxylated PVPs showed higher renal accumulation than sulfonated PVPs, and about 30% of the administered dose of 20%-carboxylated PVP was observed in the kidney. In contrast, PAA (100%-carboxylated) and PVP (0%-carboxylated) showed little accumulation in the kidney. These results suggest that carboxylated PVP that contains the optimal number of carboxyl groups might exhibit the highest accumulation in the kidney. The blood levels of all polymers decreased rapidly 24 h after i.v. injection. PVP did not accumulate in the kidneys, while anionized PVPs accumulated in the kidneys over the period studied. The maximal renal levels occurred 3h after treatment and slowly declined thereafter. In particular, the level of renal 20%-carboxylated PVP was thirty-two times higher than that of PVP 24 h after i.v. injection. These results suggest that carboxylated PVP was rapidly distributed to the kidney and was gradually excreted into the urine, whereas sulfonated PVPs were quickly excreted in urine. In contrast, PVP was effectively retained in the blood and gradually excreted into the urine without concentrating in the kidneys.
Figure 6.
Tissue distribution of PVP and anionized PVP derivatives at 3h after i.v. injection in mice. Mice were intravenously injected with 125I-labeled polymers. After i.v. injection, mice were killed and the organs were collected. The radioactivity was measured by γ-counter. Mice were used in groups of five. Each value is the mean ±S.D.
Figure 6.
Tissue distribution of PVP and anionized PVP derivatives at 3h after i.v. injection in mice. Mice were intravenously injected with 125I-labeled polymers. After i.v. injection, mice were killed and the organs were collected. The radioactivity was measured by γ-counter. Mice were used in groups of five. Each value is the mean ±S.D.
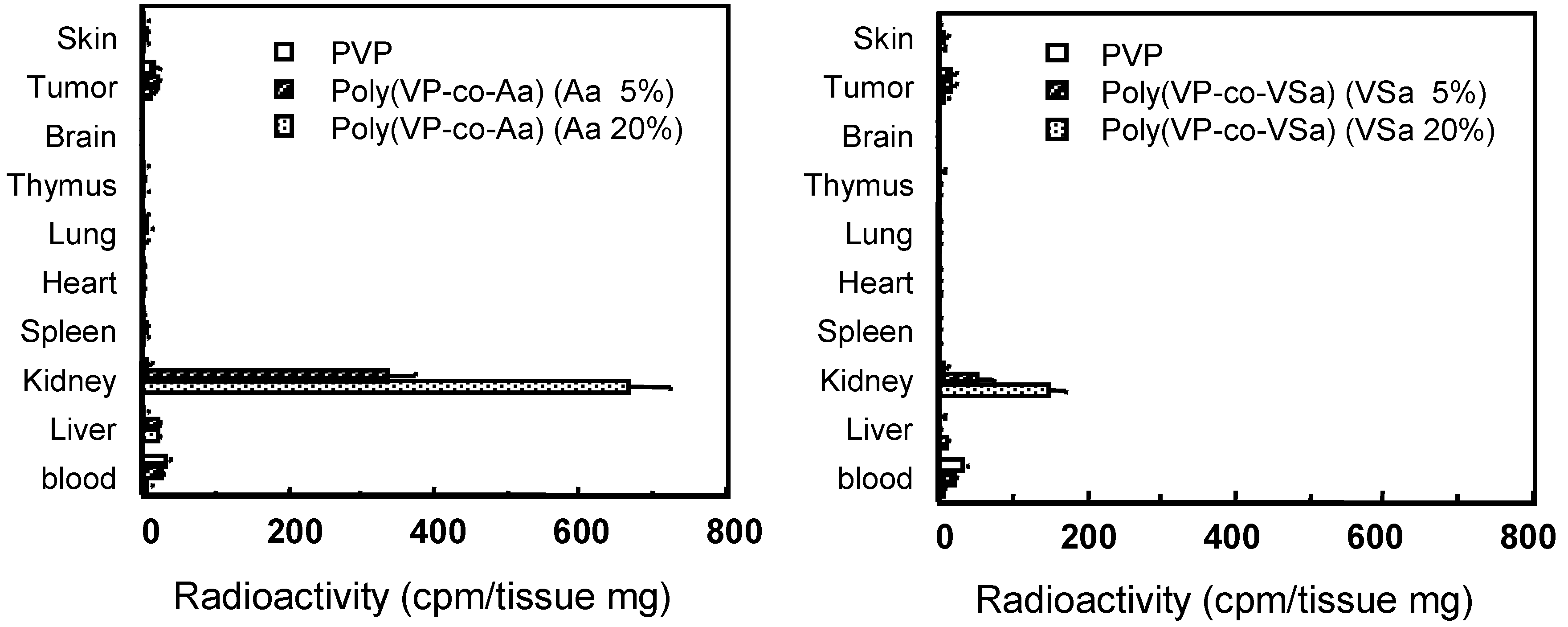
Few studies have examined the effects of carboxyl and sulfonic groups attached to a polymer on the pharmacokinetic characteristics of drug conjugates. We assessed the accumulation sites of anionized PVPs in the kidney by tissue-section analysis, and found that carboxylated PVPs were effectively localized and retained in the renal proximal tubular epithelium cells (data not shown). The mechanism of uptake of carboxylated PVPs in the proximal tubular epithelium cells in vivo is currently under investigation.
Our approach for designing polymeric modifiers involved only a few steps. In the present study, the effects of anionic charge and other physicochemical properties of the polymeric modifier were investigated. The relationship between several biological factors and physicochemical disposition is necessary to determine the biopharmaceutical characteristics of polymeric modifiers. This approach may facilitate the optimum molecular design of polymeric modifiers for drug delivery systems.
Conclusions
One of the most efficient ways of improving therapeutic potency of proteins has been to modify them with polymeric modifiers, as typified by PEG. PEGylated IFN-alpha, that has clinically been shown to have marked antiviral activity against hepatitis C. Until recently, we have also shown the usefulness of conjugation. However, clinical application of most PEGylated proteins has not yet been successful. In most cases, PEGylation occurs randomly at multiple lysine residues in the proteins, some of which may be located in or near the protein active site. The resultant PEGylated proteins are therefore heterogeneous and composed of various positional isomers with distinct specific activities. Additionally, their bioactivity is markedly lower than of the unmodified proteins. To overcome these drawbacks, we attempted to develop a strategy for site-specific PEGylation [45]. The PEGylation system in which the N terminus of the protein is specifically PEGylated after creation of lysine-deficient mutants with full bioactivity through the use of phage libraries have been successfully established. This N-terminal mono-PEGylated mutant TNF-alpha had comparable bioactivity to unmodified TNF-alpha in vitro, and other properties including plasma half-life, antitumor activity, and toxicity were greatly improved. This technology will open a new window in the area of polymer-conjugation and increase the possibility for clinical application of conjugated proteins, by adopting polymeric modifiers with useful DDS-functions as described in this review.
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research (No. 15680014 and No. 16023242) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, in part by a Health and Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare of Japan, in part by Health Sciences Research Grants for Research on Health Sciences focusing on Drug Innovation from the Japan Health Sciences Foundation, in part by Takeda Science Foundation, and in part by Senri Life Science Foundation.
References
- Glue, P.; Rouzier-Panis, R.; Raffanel, C.; Sabo, R.; Gupta, S. K.; Salfi, M.; Jacobs, S.; Clement, R. P. A dose-ranging study of pegylated interferon alfa-2b and ribavirin in chronic hepatitis C. The Hepatitis C Intervention Therapy Group. Hepatology 2000, 32, 647–53. [Google Scholar] [PubMed]
- Furman, W. L.; Strother, D.; McClain, K.; Bell, B.; Leventhal, B.; Pratt, C. B. Phase I clinical trial of recombinant human tumor necrosis factor in children with refractory solid tumors: a Pediatric Oncology Group study. J. Clin. Oncol. 1993, 11, 2205–10. [Google Scholar] [PubMed]
- Barnard, D. L. Pegasys (Hoffmann-La Roche). Curr. Opin. Investig. Drugs 2001, 2, 1530–8. [Google Scholar] [PubMed]
- Borden, E. C.; Sondel, P. M. Lymphokines and cytokines as cancer treatment. Immunotherapy realized. Cancer 1990, 65, 800–14. [Google Scholar] [PubMed]
- Waters, C. A.; Schimke, P. A.; Snider, C. E.; Itoh, K.; Smith, K. A.; Nichols, J. C.; Strom, T. B.; Murphy, J. R. Interleukin 2 receptor-targeted cytotoxicity. Receptor binding requirements for entry of a diphtheria toxin-related interleukin 2 fusion protein into cells. Eur. J. Immunol. 1990, 20, 785–91. [Google Scholar]
- Mohler, K. M.; Torrance, D. S.; Smith, C. A.; Goodwin, R. G.; Stremler, K. E.; Fung, V. P.; Madani, H.; Widmer, M. B. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J. Immunol. 1993, 151, 1548–61. [Google Scholar] [PubMed]
- Rosenberg, S. A.; Lotze, M. T.; Muul, L. M.; Chang, A. E.; Avis, F. P.; Leitman, S.; Linehan, W. M.; Robertson, C. N.; Lee, R. E.; Rubin, J. T. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–97. [Google Scholar] [PubMed]
- Gordon, M. S.; Nemunaitis, J.; Hoffman, R.; Paquette, R. L.; Rosenfeld, C.; Manfreda, S.; Isaacs, R.; Nimer, S. D. A phase I trial of recombinant human interleukin-6 in patients with myelodysplastic syndromes and thrombocytopenia. Blood 1995, 85, 3066–76. [Google Scholar] [PubMed]
- Kreitman, R. J.; Wilson, W. H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D. J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–7. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Taguchi, T.; Urushizaki, I.; Ohno, R.; Abe, O.; Furue, H.; Hattori, T.; Ichihashi, H.; Inoguchi, K.; Majima, H. Phase I study of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol. 1987, 20, 223–9. [Google Scholar] [PubMed]
- Nagata, S. Steering anti-cancer drugs away from the TRAIL. Nat. Med. 2000, 6, 502–3. [Google Scholar] [PubMed]
- Talpaz, M.; O'Brien, S.; Rose, E.; Gupta, S.; Shan, J.; Cortes, J.; Giles, F. J.; Faderl, S.; Kantarjian, H. M. Phase 1 study of polyethylene glycol formulation of interferon alpha-2B (Schering 54031) in Philadelphia chromosome-positive chronic myelogenous leukemia. Blood 2001, 98, 1708–13. [Google Scholar] [PubMed]
- Hershfield, M. S.; Buckley, R. H.; Greenberg, M. L.; Melton, A. L.; Schiff, R.; Hatem, C.; Kurtzberg, J.; Markert, M. L.; Kobayashi, R. H.; Kobayashi, A. L. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N. Engl. J. Med. 1987, 316, 589–96. [Google Scholar] [PubMed]
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv. Drug. Deliv. Rev. 2001, 46, 169–85. [Google Scholar] [CrossRef] [PubMed]
- Goodson, R. J.; Katre, N. V. Site-directed pegylation of recombinant interleukin-2 at its glycosylation site. Biotechnology (N Y) 1990, 8, 343–6. [Google Scholar]
- Chapes, S. K.; Simske, S. J.; Sonnenfeld, G.; Miller, E. S.; Zimmerman, R. J. Effects of spaceflight and PEG-IL-2 on rat physiological and immunological responses. J. Appl. Physiol. 1999, 86, 2065–76. [Google Scholar] [PubMed]
- Tsutsumi, Y.; Tsunoda, S.; Kamada, H.; Kihira, T.; Nakagawa, S.; Kaneda, Y.; Kanamori, T.; Mayumi, T. Molecular design of hybrid tumour necrosis factor-alpha. II: The molecular size of polyethylene glycol-modified tumour necrosis factor-alpha affects its anti-tumour potency. Br. J. Cancer 1996, 74, 1090–5. [Google Scholar]
- Tsutsumi, Y.; Kihira, T.; Tsunoda, S.; Kanamori, T.; Nakagawa, S.; Mayumi, T. Molecular design of hybrid tumour necrosis factor alpha with polyethylene glycol increases its anti-tumour potency. Br. J. Cancer 1995, 71, 963–8. [Google Scholar] [PubMed]
- Kaneda, Y.; Yamamoto, Y.; Kamada, H.; Tsunoda, S.; Tsutsumi, Y.; Hirano, T.; Mayumi, T. Antitumor activity of tumor necrosis factor alpha conjugated with divinyl ether and maleic anhydride copolymer on solid tumors in mice. Cancer Res. 1998, 58, 290–5. [Google Scholar]
- Tsunoda, S.; Ishikawa, T.; Yamamoto, Y.; Kamada, H.; Koizumi, K.; Matsui, J.; Tsutsumi, Y.; Hirano, T.; Mayumi, T. Enhanced antitumor potency of polyethylene glycolylated tumor necrosis factor-alpha: a novel polymer-conjugation technique with a reversible amino-protective reagent. J. Pharmacol. Exp. Ther. 1999, 290, 368–72. [Google Scholar] [PubMed]
- Kamada, H.; Tsutsumi, Y.; Tsunoda, S.; Kihira, T.; Kaneda, Y.; Yamamoto, Y.; Nakagawa, S.; Horisawa, Y.; Mayumi, T. Molecular design of conjugated tumor necrosis factor-alpha: synthesis and characteristics of polyvinyl pyrrolidone modified tumor necrosis factor-alpha. Biochem. Biophys. Res. Commun. 1999, 257, 448–53. [Google Scholar] [PubMed]
- Kamada, H.; Tsutsumi, Y.; Yamamoto, Y.; Kihira, T.; Kaneda, Y.; Mu, Y.; Kodaira, H.; Tsunoda, S. I.; Nakagawa, S.; Mayumi, T. Antitumor activity of tumor necrosis factor-alpha conjugated with polyvinylpyrrolidone on solid tumors in mice. Cancer Res. 2000, 60, 6416–20. [Google Scholar] [PubMed]
- Kodaira, H.; Tsutsumi, Y.; Yoshioka, Y.; Kamada, H.; Kaneda, Y.; Yamamoto, Y.; Tsunoda, S.; Okamoto, T.; Mukai, Y.; Shibata, H.; Nakagawa, S.; Mayumi, T. The targeting of anionized polyvinylpyrrolidone to the renal system. Biomaterials 2004, 25, 4309–15. [Google Scholar] [CrossRef]
- Debs, R. J.; Fuchs, H. J.; Philip, R.; Brunette, E. N.; Duzgunes, N.; Shellito, J. E.; Liggitt, D.; Patton, J. R. Immunomodulatory and toxic effects of free and liposome-encapsulated tumor necrosis factor alpha in rats. Cancer Res. 1990, 50, 375–80. [Google Scholar] [PubMed]
- Nobuhara, M.; Kanamori, T.; Ashida, Y.; Ogino, H.; Horisawa, Y.; Nakayama, K.; Asami, T.; Iketani, M.; Noda, K.; Andoh, S. The inhibition of neoplastic cell proliferation with human natural tumor necrosis factor. Jpn. J. Cancer Res. 1987, 78, 193–201. [Google Scholar] [PubMed]
- Moritz, T.; Niederle, N.; Baumann, J.; May, D.; Kurschel, E.; Osieka, R.; Kempeni, J.; Schlick, E.; Schmidt, C. G. Phase I study of recombinant human tumor necrosis factor alpha in advanced malignant disease. Cancer Immunol. Immunother. 1989, 29, 144–50. [Google Scholar]
- Noguchi, K.; Inagawa, H.; Tsuji, Y.; Morikawa, A.; Mizuno, D.; Soma, G. Antitumor activity of a novel chimera tumor necrosis factor (TNF-STH) constructed by connecting rTNF-S with thymosin beta 4 against murine syngeneic tumors. J. Immunother. 1991, 10, 105–11. [Google Scholar] [PubMed]
- Blick, M.; Sherwin, S. A.; Rosenblum, M.; Gutterman, J. Phase I study of recombinant tumor necrosis factor in cancer patients. Cancer Res. 1987, 47, 2986–9. [Google Scholar] [PubMed]
- Hill, R. J.; Warren, M. K.; Stenberg, P.; Levin, J.; Corash, L.; Drummond, R.; Baker, G.; Levin, F.; Mok, Y. Stimulation of megakaryocytopoiesis in mice by human recombinant interleukin-6. Blood 1991, 77, 42–8. [Google Scholar] [PubMed]
- Ishibashi, T.; Kimura, H.; Shikama, Y.; Uchida, T.; Kariyone, S.; Hirano, T.; Kishimoto, T.; Takatsuki, F.; Akiyama, Y. Interleukin-6 is a potent thrombopoietic factor in vivo in mice. Blood. 1989, 74, 1241–4. [Google Scholar] [PubMed]
- Zeidler, C.; Kanz, L.; Hurkuck, F.; Rittmann, K. L.; Wildfang, I.; Kadoya, T.; Mikayama, T.; Souza, L.; Welte, K. In vivo effects of interleukin-6 on thrombopoiesis in healthy and irradiated primates. Blood 1992, 80, 2740–5. [Google Scholar] [PubMed]
- Navarro, S.; Debili, N.; Le Couedic, J. P.; Klein, B.; Breton-Gorius, J.; Doly, J.; Vainchenker, W. Interleukin-6 and its receptor are expressed by human megakaryocytes: in vitro effects on proliferation and endoreplication. Blood 1991, 77, 461–71. [Google Scholar] [PubMed]
- Taga, T.; Kishimoto, T. Role of a two-chain IL-6 receptor system in immune and hematopoietic cell regulation. Crit. Rev. Immunol. 1992, 11, 265–80. [Google Scholar]
- Castell, J. V.; Geiger, T.; Gross, V.; Andus, T.; Walter, E.; Hirano, T.; Kishimoto, T.; Heinrich, P. C. Plasma clearance, organ distribution and target cells of interleukin-6/hepatocyte-stimulating factor in the rat. Eur. J. Biochem. 1988, 177, 357–61. [Google Scholar] [PubMed]
- Suematsu, S.; Matsuda, T.; Aozasa, K.; Akira, S.; Nakano, N.; Ohno, S.; Miyazaki, J.; Yamamura, K.; Hirano, T.; Kishimoto, T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7547–51. [Google Scholar] [PubMed]
- Weber, J.; Yang, J. C.; Topalian, S. L.; Parkinson, D. R.; Schwartzentruber, D. S.; Ettinghausen, S. E.; Gunn, H.; Mixon, A.; Kim, H.; Cole, D. Phase I trial of subcutaneous interleukin-6 in patients with advanced malignancies. J. Clin. Oncol. 1993, 11, 499–506. [Google Scholar] [PubMed]
- Delgado, C.; Francis, G. E.; Fisher, D. The uses and properties of PEG-linked proteins. Crit. Rev. Ther. Drug. Carrier. Syst. 1992, 9, 249–304. [Google Scholar]
- Inoue, M.; Ebashi, I.; Watanabe, N.; Morino, Y. Synthesis of a superoxide dismutase derivative that circulates bound to albumin and accumulates in tissues whose pH is decreased. Biochemistry. 1989, 28, 6619–24. [Google Scholar] [PubMed]
- Nagasaki, Y.; Iijima, M.; Kato, M.; Kataoka, K. Primary amino-terminal heterobifunctional poly(ethylene oxide). Facile synthesis of poly(ethylene oxide) with a primary amino group at one end and a hydroxyl group at the other end. Bioconjug. Chem. 1995, 6, 702–4. [Google Scholar] [PubMed]
- Kaneda, Y.; Tsutsumi, Y.; Yoshioka, Y.; Kamada, H.; Yamamoto, Y.; Kodaira, H.; Tsunoda, S.; Okamoto, T.; Mukai, Y.; Shibata, H.; Nakagawa, S.; Mayumi, T. The use of PVP as a polymeric carrier to improve the plasma half-life of drugs. Biomaterials 2004, 25, 3259–66. [Google Scholar] [PubMed]
- Tsunoda, S.; Kamada, H.; Yamamoto, Y.; Ishikawa, T.; Matsui, J.; Koizumi, K.; Kaneda, Y.; Tsutsumi, Y.; Ohsugi, Y.; Hirano, T.; Mayumi, T. Molecular design of polyvinyl- pyrrolidone-conjugated interleukin-6 for enhancement of in vivo thrombopoietic activity in mice. J. Control. Release 2000, 68, 335–41. [Google Scholar] [PubMed]
- Simionescu, N. Cellular aspects of transcapillary exchange. Physiol. Rev. 1983, 63, 1536–79. [Google Scholar] [PubMed]
- Chang, R. L.; Deen, W. M.; Robertson, C. R.; Brenner, B. M. Permselectivity of the glomerular capillary wall: III. Restricted transport of polyanions. Kidney Int. 1975, 8, 212–8. [Google Scholar] [PubMed]
- Takakura, Y.; Fujita, T.; Hashida, M.; Sezaki, H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm. Res. 1990, 7, 339–46. [Google Scholar] [PubMed]
- Yamamoto, Y.; Tsutsumi, Y.; Yoshioka, Y.; Nishibata, T.; Kobayashi, K.; Okamoto, T.; Mukai, Y.; Shimizu, T.; Nakagawa, S.; Nagata, S.; Mayumi, T. Site-specific PEGylation of a lysine-deficient TNF-alpha with full bioactivity. Nat. Biotechnol. 2003, 21, 546–52. [Google Scholar] [PubMed]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Shibata, H.; Nakagawa, S.; Tsutsumi, Y. Optimization of Protein Therapies by Polymer-Conjugation as an Effective DDS. Molecules 2005, 10, 162-180. https://doi.org/10.3390/10010162
AMA Style
Shibata H, Nakagawa S, Tsutsumi Y. Optimization of Protein Therapies by Polymer-Conjugation as an Effective DDS. Molecules. 2005; 10(1):162-180. https://doi.org/10.3390/10010162
Chicago/Turabian StyleShibata, Hiroko, Shinsaku Nakagawa, and Yasuo Tsutsumi. 2005. "Optimization of Protein Therapies by Polymer-Conjugation as an Effective DDS" Molecules 10, no. 1: 162-180. https://doi.org/10.3390/10010162