Fragrance Release from the Surface of Branched Poly (Amide) S
by
F. Aulenta
1,
M. Drew
1,
A. Foster
2,
W. Hayes
1,*, 
S. Rannard
2,
D. Thornthwaite
2 and
T. Youngs
1 1
School of Chemistry, The University of Reading, UK
2
Unilever Research and Development, Port Sunlight Laboratory, UK
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(1), 81-97; https://doi.org/10.3390/10010081
Submission received: 14 May 2004
/
Accepted: 29 June 2005
/
Published: 31 January 2005
(This article belongs to the Special Issue Macromolecules Applied to Pharmaceutical Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Enzymes are powerful tools in organic synthesis that are able to catalyse a wide variety of selective chemical transformations under mild and environmentally friendly conditions. Enzymes such as the lipases have also found applications in the synthesis and degradation of polymeric materials. However, the use of these natural catalysts in the synthesis and the post-synthetic modification of dendrimers and hyperbranched molecules is an application of chemistry yet to be explored extensively. In this study the use of two hydrolytic enzymes, a lipase from Candida cylindracea and a cutinase from Fusarium solani pisii, were investigated in the selective cleavage of ester groups situated on the peripheral layer of two families of branched polyamides. These branched polyamides were conjugated to simple fragrances citronellol and l-menthol via ester linkages. Hydrolysis of the ester linkage between the fragrances and the branched polyamide support was carried out in aqueous buffered systems at slightly basic pH values under the optimum operative conditions for the enzymes used. These preliminary qualitative investigations revealed that partial cleavage of the ester functionalities from the branched polyamide support had occurred. However, the ability of the enzymes to interact with the substrates decreased considerably as the branching density, the rigidity of the structure and the bulkiness of the polyamide-fragrance conjugates increased.
Introduction
The majority of the enzymes operate at physiological temperatures, in neutral aqueous media, and in the absence of substrate functional group protection [1]. Furthermore, in many cases enzymes display chemo-, regio- and enantioselectivity, and possess high turnover values, thereby rendering these natural catalysts very appealing, especially in the pharmaceutical and agrochemical industries [2]. The use of enzymes as catalysts has also been exploited in areas such as asymmetric synthesis [3]. Esterases, lipases and proteases – all hydrolytic biocatalysts, have been employed extensively in the preparation of enantiopure compounds from racemic mixtures, prochiral and meso compounds [4]. However, enzymes are also used frequently in the protection/deprotection of sugars and peptides, where selective masking of α-amino groups, carboxy groups and various side chains represent the most important problems in peptide chemistry [5]. Lipases are a class of enzymes employed frequently in organic synthesis and catalyse the hydrolysis of esters in aqueous medium and the esterification of carboxylic acids in organic solvents [6]. Numerous lipases have also been used frequently in the synthesis, transformation and biodegradation studies of polymeric materials [7].
A notable example of enzyme-catalysed polymeric degradation has been described by Hayashi and co-workers. The lipase from Pseudomonas fluorescence was used in the in vitro degradation of poly(hexano-6-lactone) (PLC) fibres [8]. Seebach and co-workers reported subsequently the enzyme-promoted degradation of ester moieties from a dendritic surface [9] and this approach has great potential in material science and drug delivery. Indeed, the use of enzymes for the selective post-synthetic modification of hyperbranched molecules is still a branch of chemistry yet to be exploited fully.
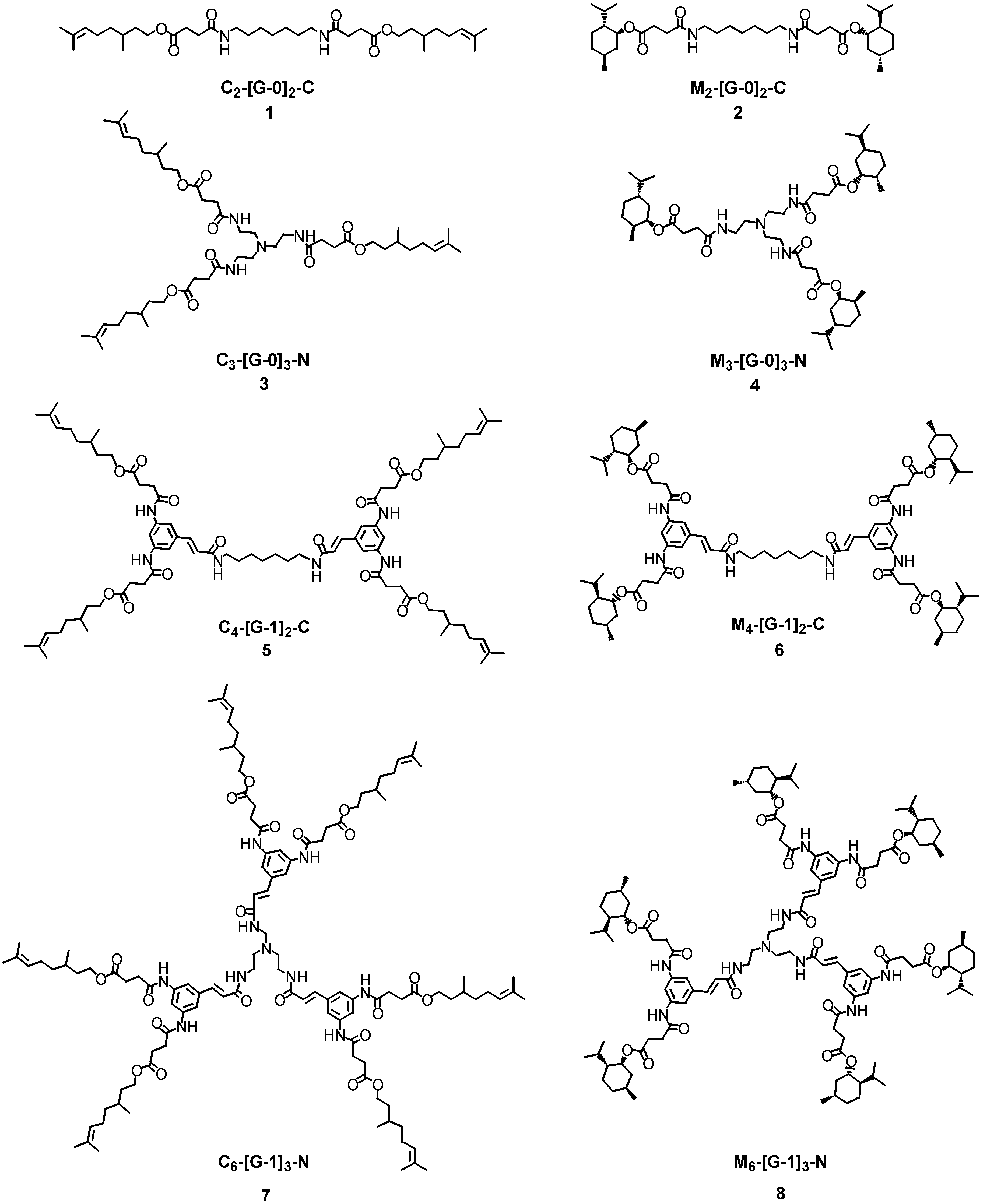
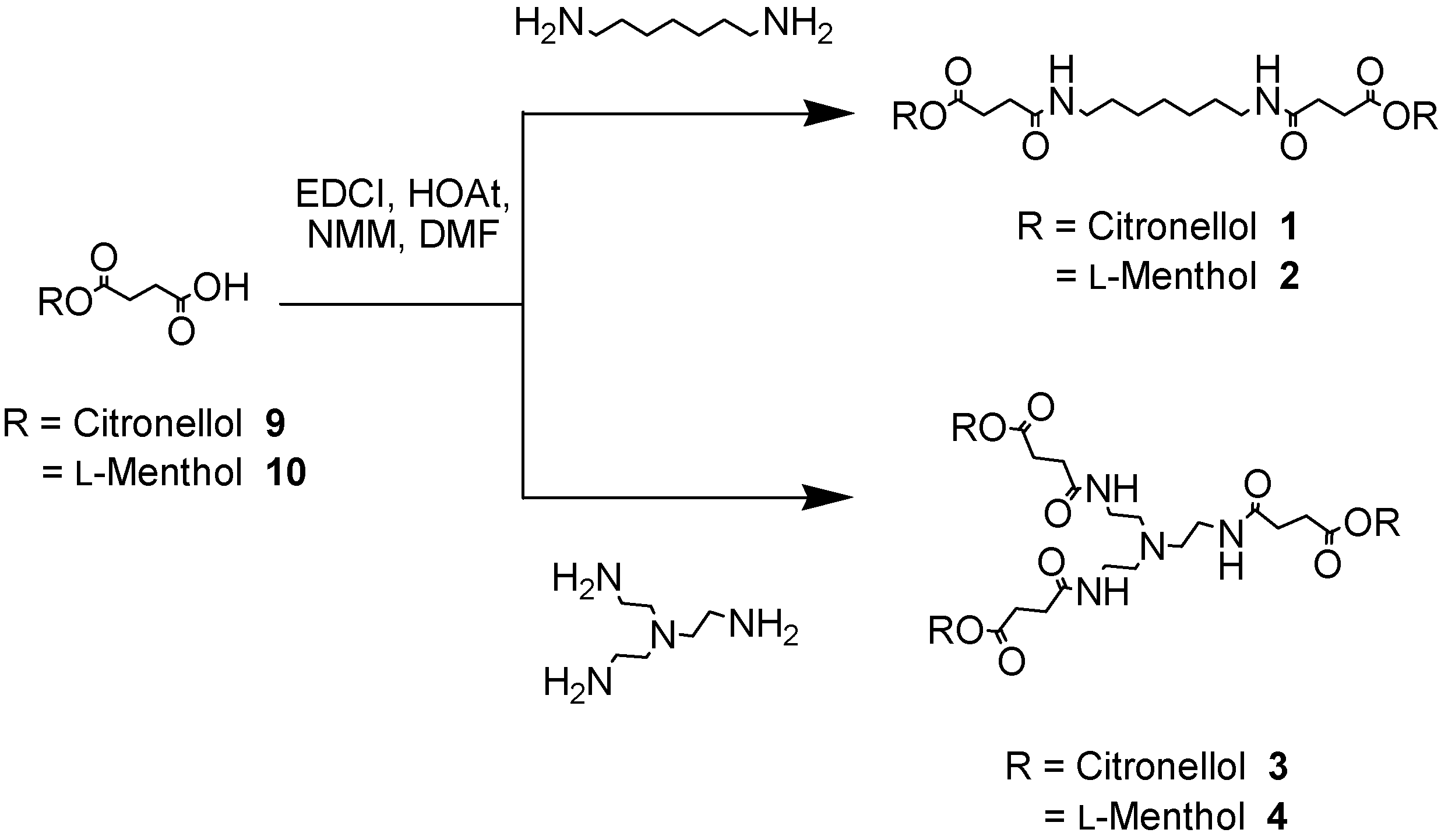
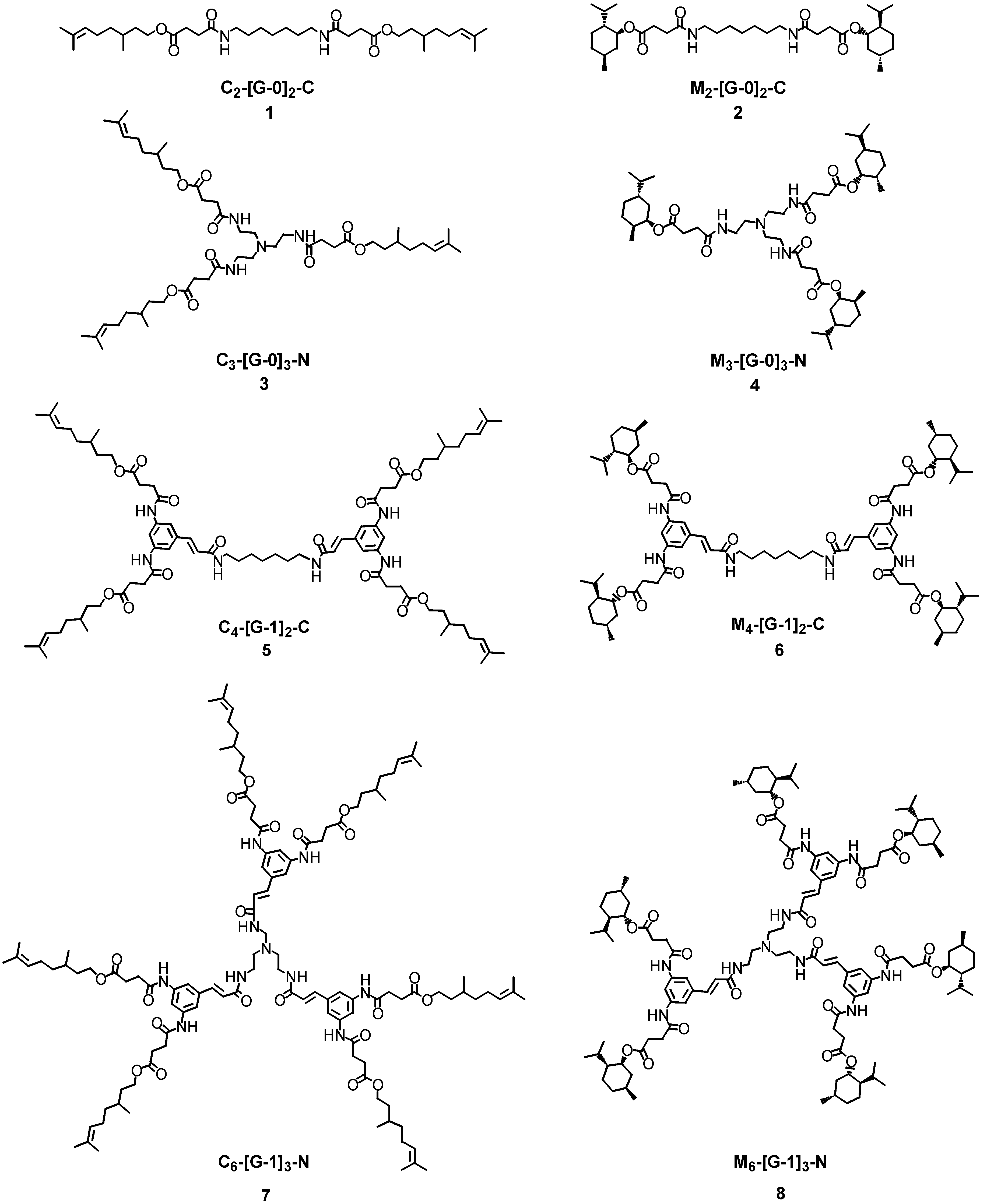
The enzyme mediated selective hydrolysis of ester functionalities on the external layers of simple linear and branched polyamides architectures (see 1-8 in Figure 1) is described in this paper. Two different ester types were conjugated onto the surfaces of the polyamides – either esters derived from a primary alcohol, citronellol or from a secondary alcohol, l-menthol. It was hoped that the use of different esters would lead to different hydrolytic profiles and thus provide information upon the enzyme selectivity and specificity. In order to modify selectively the outer layer of the hyperbranched molecules, it was decided to employ branched block-copolymers featuring polyamide functionalities in the interior and ester linkages on the periphery. It was then envisaged to use hydrolytic enzymes that are selective towards ester-type bonds, such as lipases and cutinases. The hydrolytic cleavage of the ester bonds was performed under the optimum operative conditions for the enzymes – generally aqueous buffered systems at almost neutral pH and the alcohol release profiles were monitored by reverse phase high performance liquid chromatography (RP-HPLC), gas chromatography (GC) and MALDI-TOF mass spectrometry. Mass spectrometry was employed in order to identify possible degradation products and to investigate the hydrolytic processes. The nomenclature used in this report to describe the dendrons and dendrimers was first proposed by Fréchet [10], whereby the nature and the number of peripheral functional groups are followed by the generation number (generally reported in squared brackets) and finally by the nature of functional group at the focal point or at the core. The perfume molecules resident on the outer shell of the dendrons/dendrimers are described using the abbreviations C and M for citronellol and l-menthol, respectively.
Figure 1.
Linear and Branched Polyamides 1-8 Bearing Ester Functionalities on the External Surface.
Results and Discussion
Synthesis
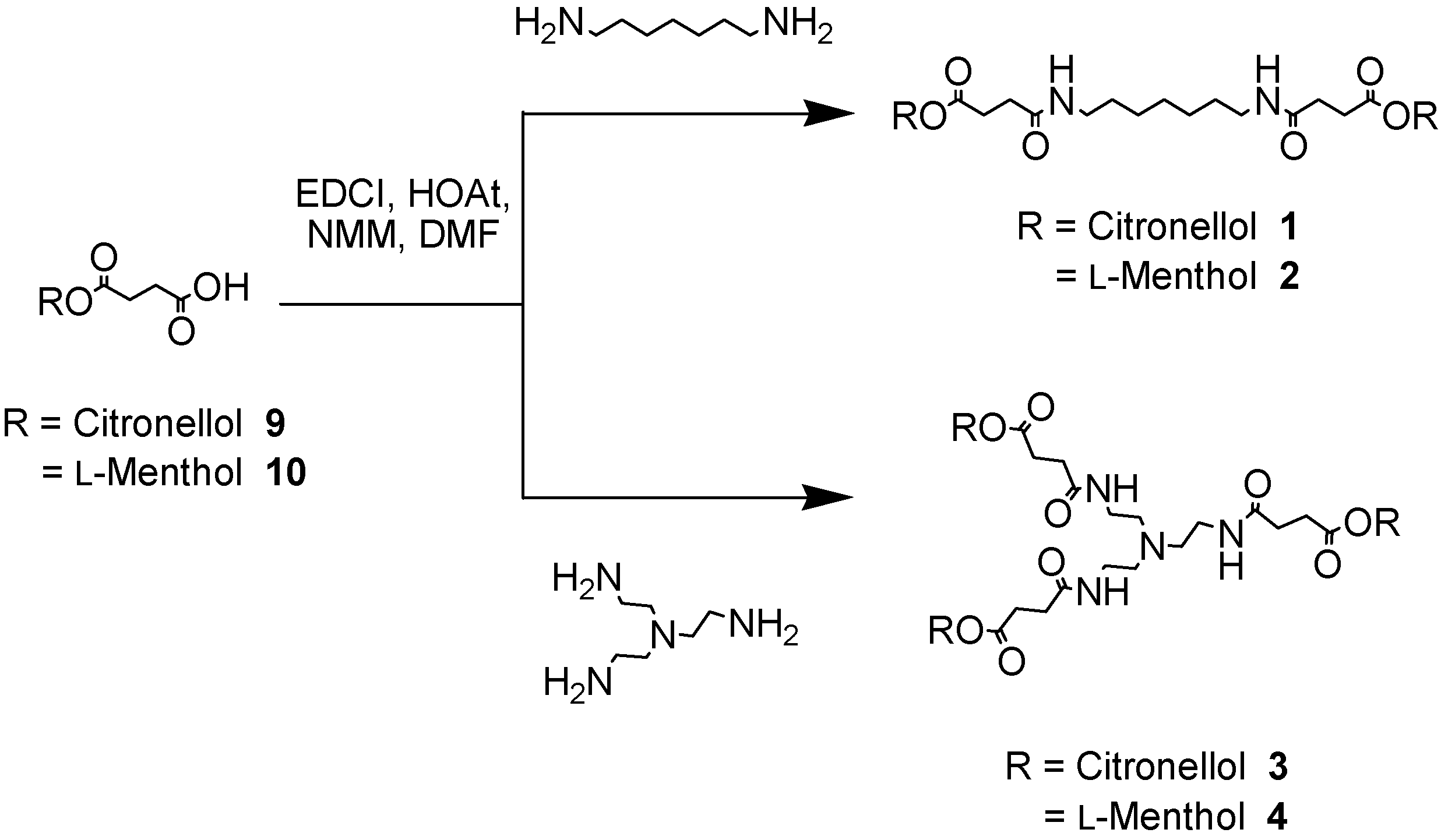
The linear and branched polyamides 1-8 bearing the ester functionalities on the external surfaces were synthesised in a convergent [11] fashion starting from the corresponding primary or secondary alcohols, respectively [12]. Succinic anhydride was ring opened under basic conditions [13] using citronellol and l-menthol, leading to the production of the carboxyester derivatives 9 as a white solid (in 95% yield) and 10 as a transparent oil (in 90% yield), respectively. The ester-carboxylic acid derivatives 9 and 10 were then coupled to the ‘core’ amine units such as 1,7-diaminoheptane or tris-(2-ethylamino)amine to afford the desired polyamides 1-4 featuring an increasing number of ester moieties (Scheme 1).
Scheme 1.
Synthesis of simple and branched polyamides (1-4)
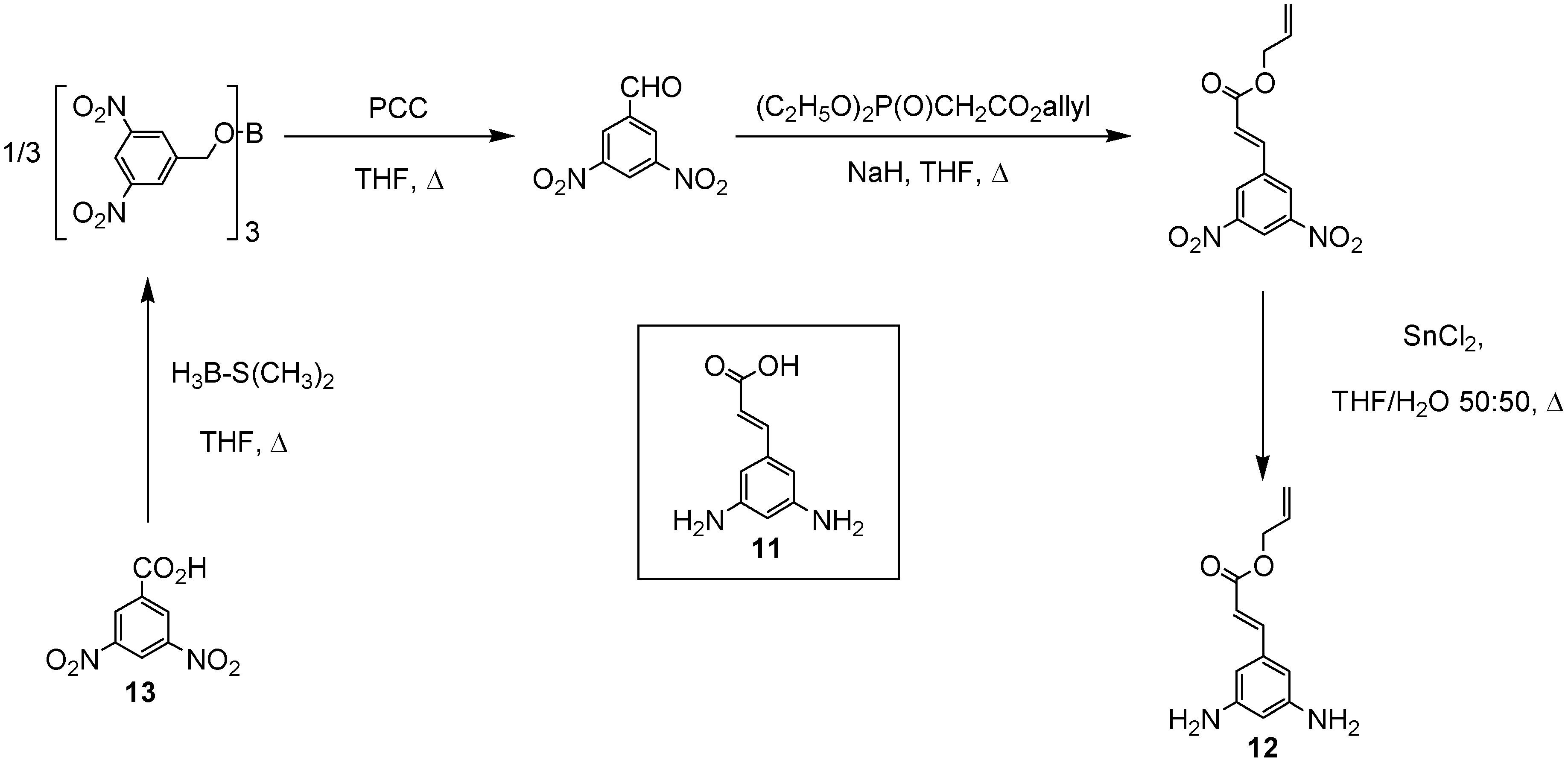
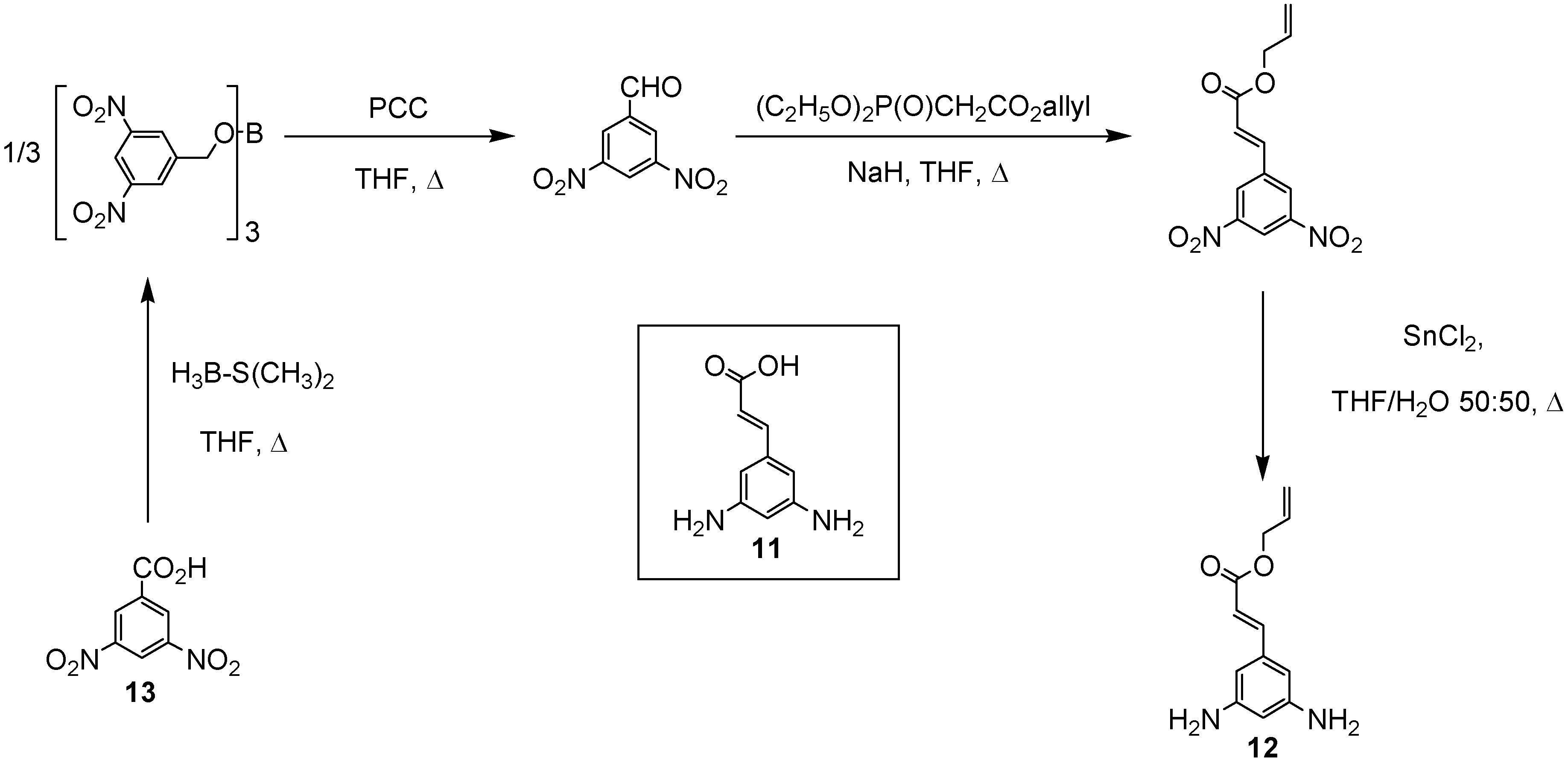
In order to increase the number of ester groups on the core units described above, the cinnamic acid derivative 3,5-diaminocinnamic acid 11 [12] was used as a branching moiety in the construction of the branched polyamides. The selection of the convergent approach [11] to the desired branched polyamides via selective coupling between the orthogonal amino moieties of 3,5-diaminocinnamic acid 11 and the carboxylic acid functionalities of the l-menthol and citronellol esters 9 and 10 necessitated the use of protecting group chemistries. Therefore, the allyl derivative of 3,5-diamino-cinnamic acid 12 was synthesised in four steps from the commercially available 3,5-dinitrobenzoic acid 13 (Scheme 2) in good yield.
Scheme 2.
Synthesis of 3,5-diaminocinnamic acid allyl ester (12)
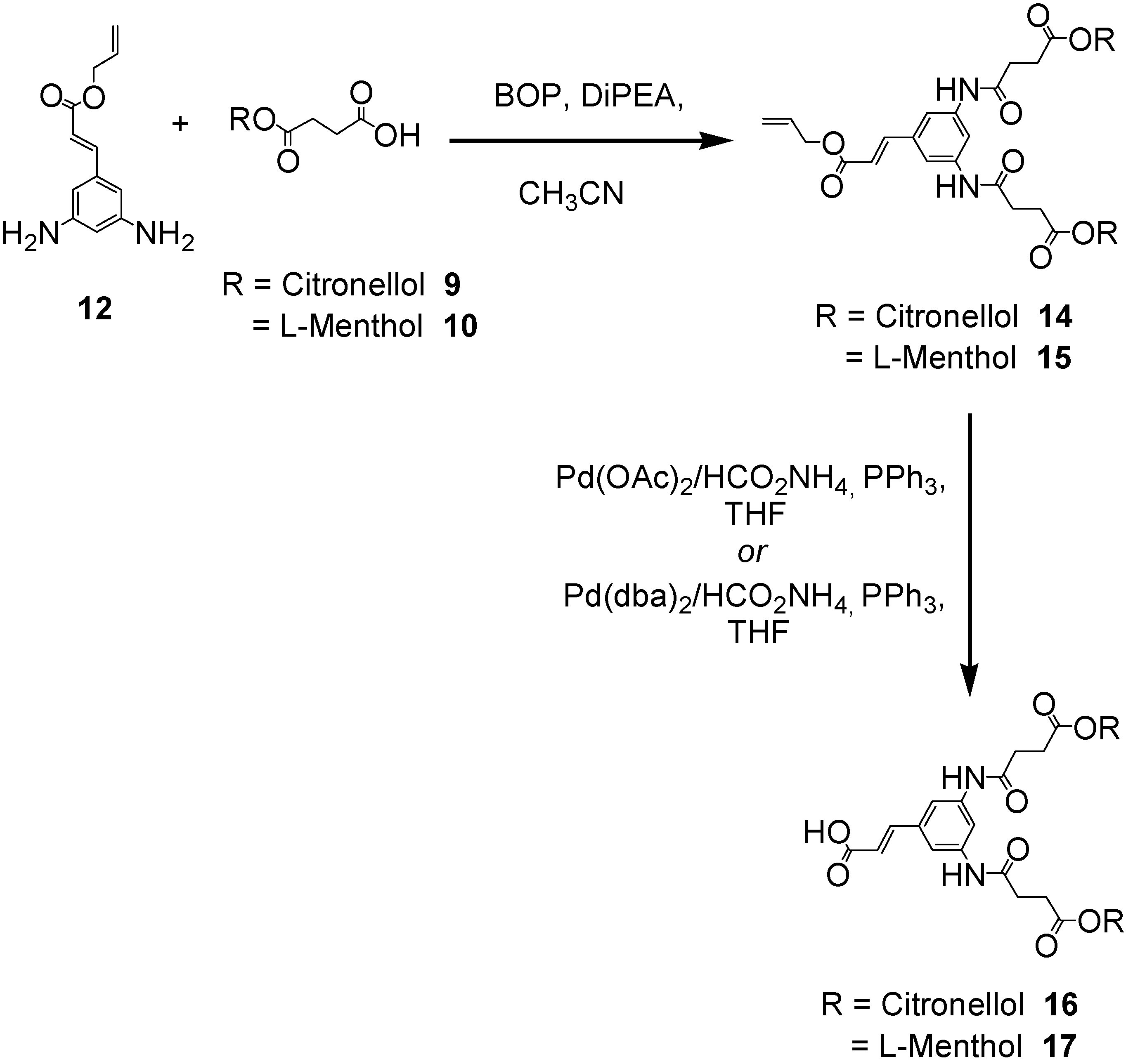
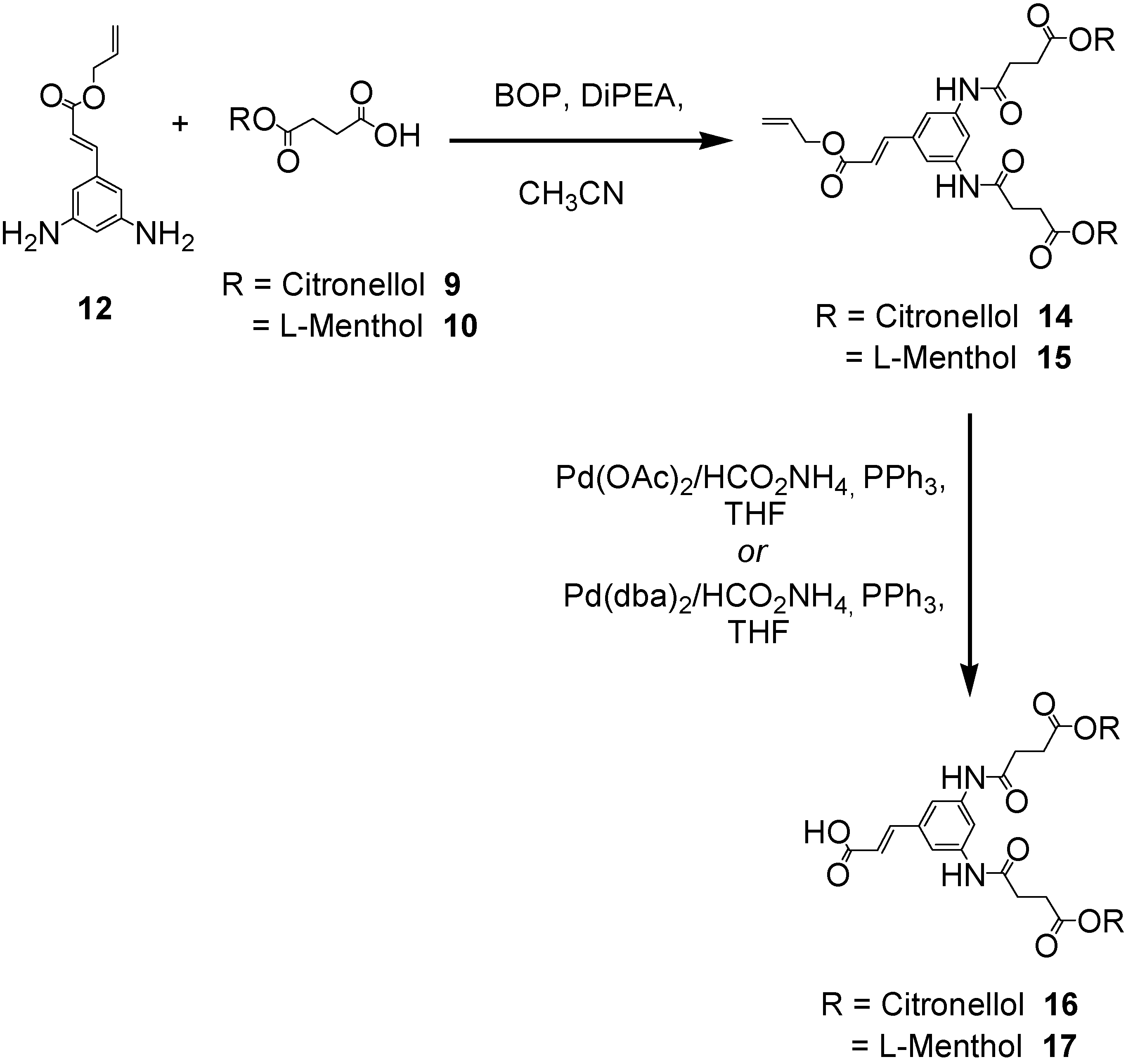
The first generation polyamide dendrons C2-[G-1]-CO2allyl (14) and M2-[G-1]-CO2allyl (15) were constructed in excellent yields (90-98%) using either 1-ethyl-3-(3’-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) and 1–hydroxy-7-azabenzotriazole (HOAt) or benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) coupling reagents (Scheme 3) [14]. Cleavage of the allyl ester at the dendrons’ focal point required very mild deprotection conditions (either Pd(OAc)2, Pd(dba)2, Pd(PPh3)4 in conjunction with ammonium formate) in order to prevent detrimental decomposition of the terpene ester linkages and to afford acceptable yields (64-74%) of the desired carboxylic acid derivatives 16 and 17.
Scheme 3.
Synthesis of the first generation polyamide dendrons featuring either citronellol or l-menthol at the peripheral surface.
Scheme 3.
Synthesis of the first generation polyamide dendrons featuring either citronellol or l-menthol at the peripheral surface.
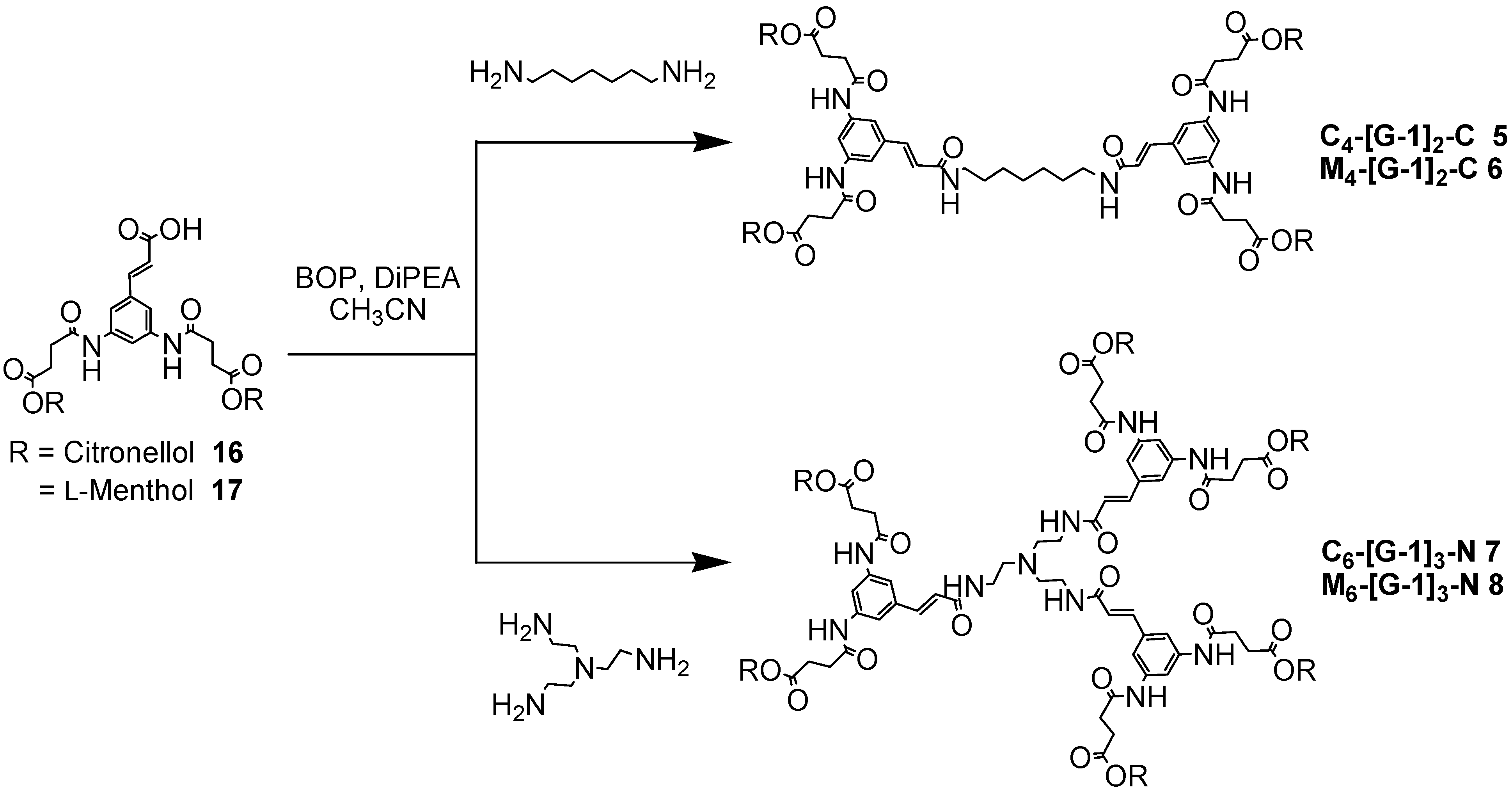
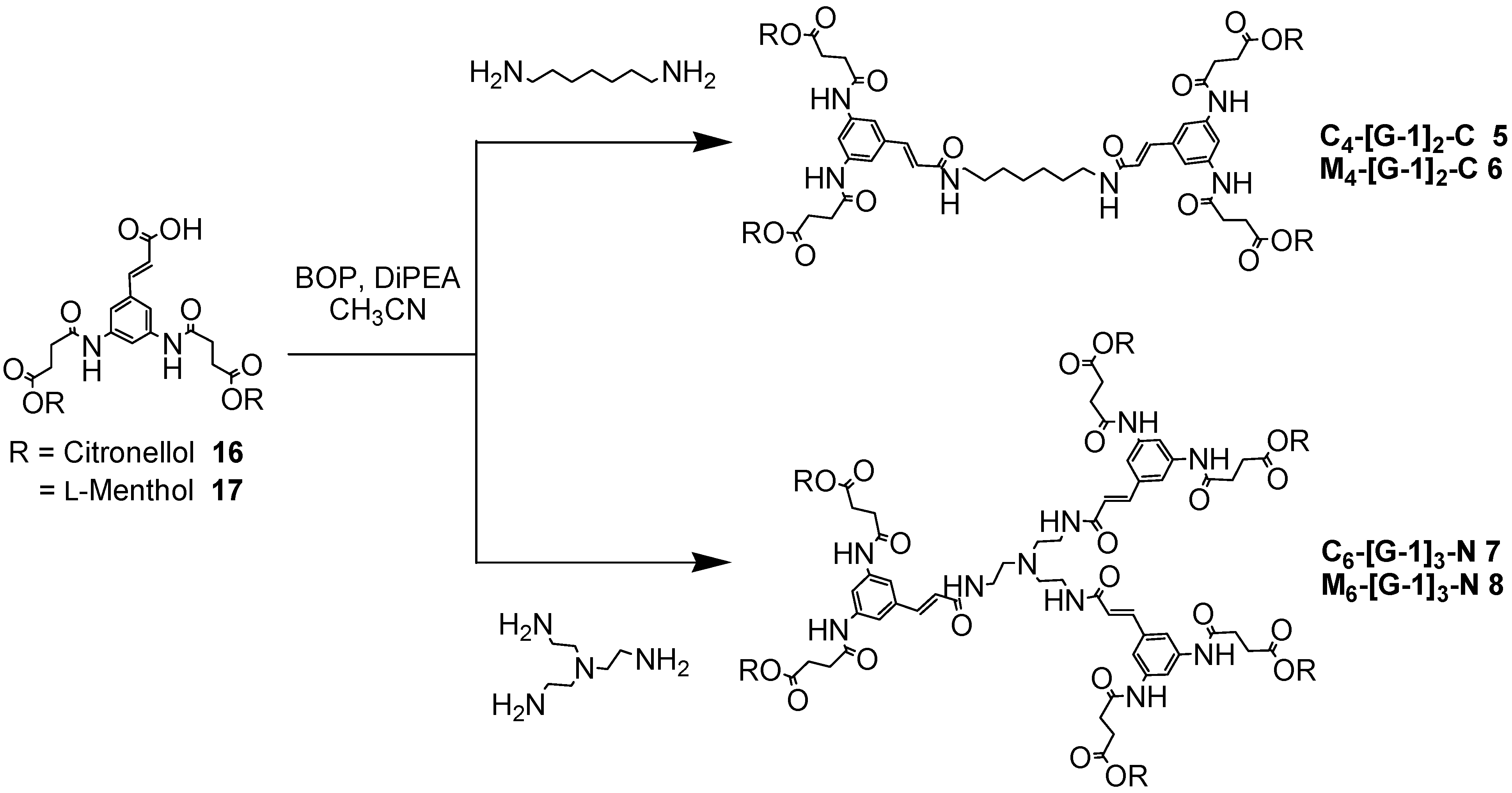
The dendrimers were constructed from [G-0] to [G-2] by coupling each dendron to either bifunctional or trifunctional central core units. Synthesis of the first generation dendrimers was carried out initially using EDCI/HOAt as the coupling reagent system, but the yields were very poor. Use of BOP afforded M4-[G-1]2-C (6) as a white solid in excellent yield (96%) and M6-[G-1]3-N (8) as pale yellow solids in moderate yield (48%) over a prolonged reaction time (48 hours). The BOP reagent was also used in the synthesis of C4-[G-1]2-C (5) and C6-[G-1]3-N (7). These dendrimers were obtained after purification by column chromatography as pale yellow solids in acceptable yields (98% and 28%, respectively), in agreement with the results obtained for the series of l-menthol first generation dendrimers (Scheme 4).
Scheme 4.
Synthesis of the branched polyamides 5-8.
Hydrolysis Studies
Preliminary studies of alcohol release under enzymatic hydrolysis were conducted on the ester groups situated on the peripheral layer of the linear and branched polyamides 1-8 with lipase from Candida cylindracea in an aqueous solution buffered to pH 7.2 [15]. The sampling was carried out over variable times (between 40 and 96 hours) and the alcohol released from the peripheral surface of the polyamides was extracted from the aqueous phases with an organic solvent (CHCl3) and its concentration monitored by RP-HPLC or GC analyses. The extracts were also analysed by MALDI-TOF mass spectrometry, from which it was possible to identify several of the major hydrolytic products. For example, when C2-[G-0]2-C (1) was subjected to the hydrolysis in the presence of the lipase an increase in the concentration of citronellol (~15%) was exhibited. However, a similar trend was also observed during the hydrolysis of 1 carried out without the lipase. This result may suggest that a slow hydrolytic process occurred as a consequence of the slightly basic conditions of the solution, and the enzyme did not participate in the catalysis of the process. However, the MALDI-TOF mass spectra of the samples extracted from the reaction mixtures did not reveal the presence of species that could be attributable to products of the hydrolysis. Therefore, it was not possible to confirm the occurrence of the hydrolytic process in this case.
In contrast, unambiguous results were obtained from the hydrolysis of M2-[G-0]2-C (2), where cleavage of the ester linkages was not observed. This result was not unexpected since compound 2 features esters that are derived from a secondary alcohol, and hence more difficult to cleave either under basic conditions or by enzymatic hydrolysis.
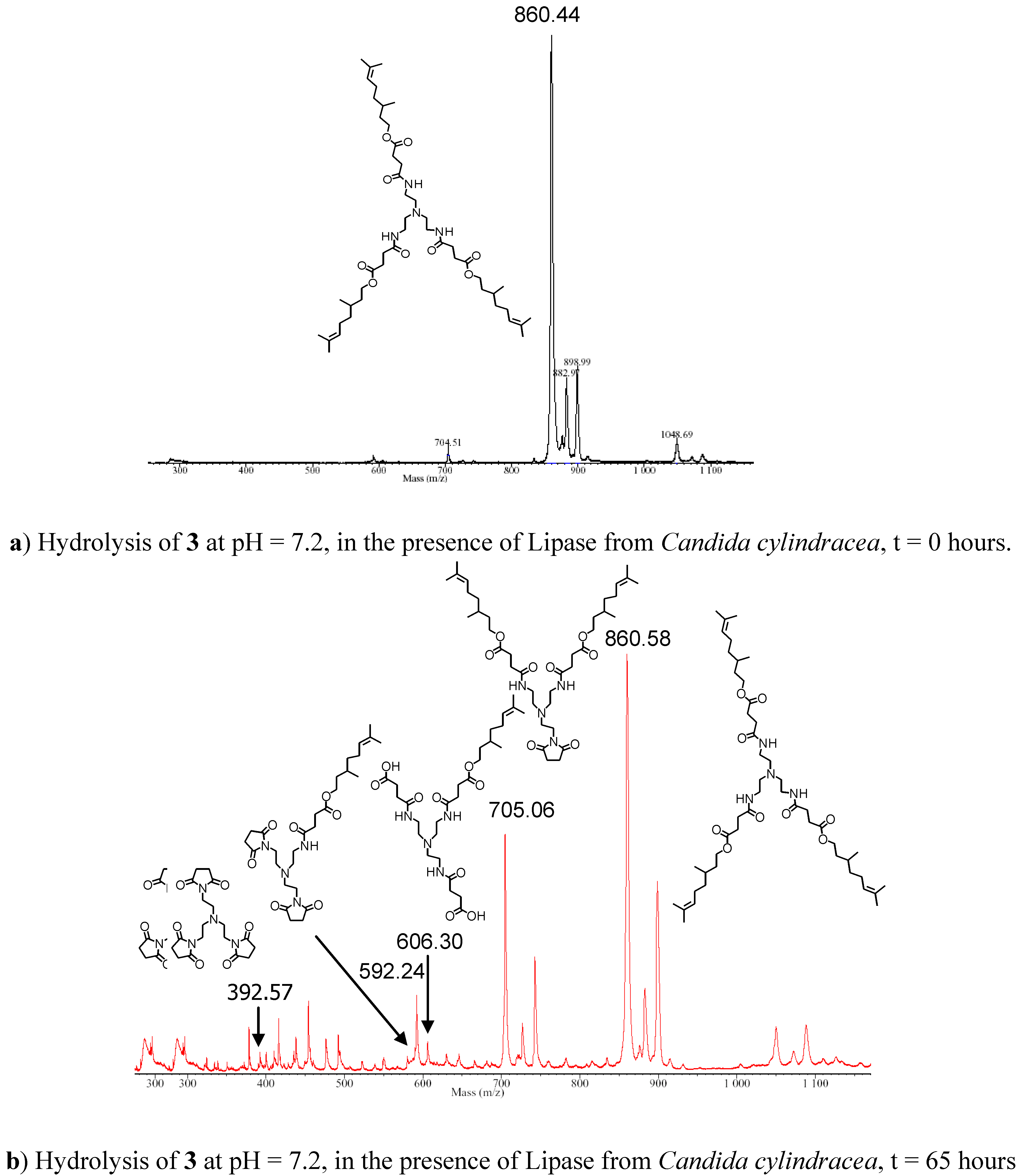
More interesting results were obtained from the hydrolysis studies of C3-[G-0]3-N (3). This compound was characterised by the presence of three citronellol moieties bound to the aliphatic polyamide structure via a succinate spacer. Furthermore, a tertiary amine functionality was present in the core unit of the molecule. When 3 was subjected to the hydrolytic conditions in the presence of lipase from Candida cylindracea progressive increase in the concentration of citronellol (~50%) was revealed by the RP-HPLC analysis. Although a certain degree of ester cleavage was also observed in the absence of enzyme (ca. 25 %), in this case a significant difference between the enzymatic hydrolysis and that performed without the enzyme was evident. MALDI-TOF mass spectrometric analysis of the samples during the hydrolysis study revealed important information about the hydrolytic process. In both reactions (the hydrolysis in the presence and absence of the enzyme) the appearance of two peaks corresponding to the loss of one molecule of citronellol (MH+=704 Da) and of two citronellol units (MH+=548 Da, or as the MK+=589 Da), was observed, which suggested formation of cyclic imides (Figure 2). The peak at 704 Da increased throughout the reaction, while the peak corresponding to the loss of two molecules of citronellol (592 Da) appeared at the initial stage of the hydrolytic process, but its relative intensity did not increase considerably as the hydrolysis proceeded. However, from the mass spectrometric analysis, it was not possible to obtain any correlation between peak intensities and concentration of the species present, therefore, it was only possible to suggest that the main hydrolytic pathway involved the release of a single molecule of citronellol per molecule of starting material C3-[G-0]3-N (3), and formation of a cyclic succinimide followed by subsequent release from the monoimide species. The other peaks detected in the spectra were attributed to the matrix used (4-hydroxy-α-cyanocinnamic acid), its aggregates and fragments, as confirmed by the spectrum obtained by the analysis of the matrix on its own recorded under identical analytical conditions. Formation of succinimides from succinates under neutral and basic conditions are well-documented in the literature [16]. For example, Matsumoto et al. [17] have reported recently the use of succinyl ester derivatives as an easily hydrolysable spacer for the conjugation of a new class of prodrug form of an HIV protease inhibitor. The esters of succinamic acids are known to undergo rapid hydrolysis via succinimide formation under mild alkaline conditions [18].
Figure 2.
MALDI-TOF Mass Spectra of the hydrolysis of C3-[G-0]3-N (3) recorded at (a) time = 0 and (b) after 65 Hours.
Figure 2.
MALDI-TOF Mass Spectra of the hydrolysis of C3-[G-0]3-N (3) recorded at (a) time = 0 and (b) after 65 Hours.
Recently, the use of lipase from Candida cylindracea to generate amide bonds in organic solvents from the corresponding esters has been reported by Gotor and co-workers [19]. Furthermore, the lipase from Pseudomonas cepacia has been employed in the enzymatic hydrazinolysis of diesters and the synthesis of N-aminosuccinimide derivatives in organic solvents [20]. The principle of ring closure with consequent elimination of an alcohol as a leaving group has also been embraced for the development of controlled release of fragrance alcohols from poly(propyleneimine) dendrimers [21]. In this case, fragrance release was based upon proton abstraction from a carbamoyl moiety by a basic buffer system. The intermediate nucleophile generated then underwent an intramolecular ring closure on the neighbouring ester moiety to release the desired fragrance and generate a cyclic imide by-product. The cyclisation occurred rapidly in slightly alkaline conditions and represented the driving force for the efficient release of even tertiary alcohols. In the last example, the efficiency of the cyclisation was promoted by the presence of a rigid benzene ring that held the two reactive components in close proximity with the conformation most appropriate to afford efficient imide ring closure.
In the current study, the linker used to conjugate the alcohol unit to the dendritic surface was a flexible succinate chain. Therefore, it is possible to assume that the pH-promoted cyclisation was not favoured as in the study reported by Hermann and co-workers [21], and occurred at a slower rate. This explanation does not contradict the different hydrolytic profiles exhibited by C3-[G-0]3-N (3) in the presence and absence of the lipase. It is proposed that in this case (hydrolysis of 3 in the buffered system) the enzyme simply accelerated a process that was already occurring. It is also proposed that the presence of the tertiary amine at the core unit of the molecule participated in the de-protonation of the amide, hence promoting the cyclisation even in the absence of the enzyme. It was noticeable that the ring closing process did not occur in the case of C2-[G-0]2-C (1), which was characterised by the same succinyl ester moieties, no central tertiary amine and a longer alkyl chain as the core unit.
When the hydrolysis of M3-[G-0]3-N (4) was investigated, release of the secondary alcohol was not observed either in the presence or absence of the enzyme. This result revealed: – i) the lack of interactions between the enzyme and the substrate, which may be a consequence of the rigidity of the structure of the alcohol moieties, and the presence of esters of a secondary alcohol and ii) the low tendency of these esters to undergo ring closing under neutral conditions, as a consequence of the poor ability of secondary alcohols to behave as leaving group under mildly basic conditions.
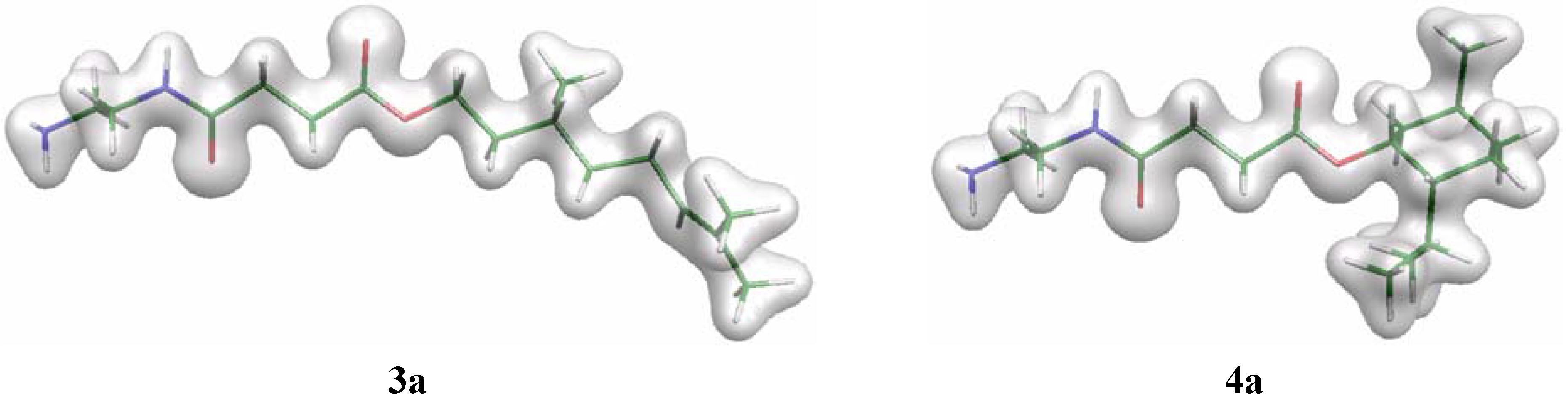
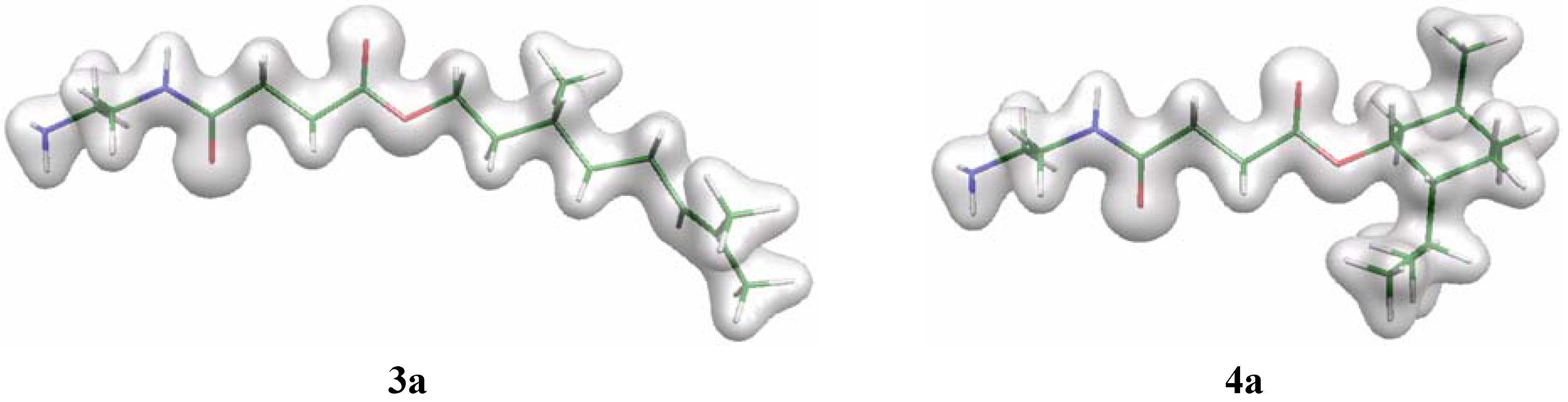
In order to examine the hydrolysis characteristics of molecules C3-[G-0]3-N (3) and M3-[G-0]3-N (4) further, preliminary computer modelling studies were performed on single ‘arms’ of the polyamides, (3a and 4a, respectively – see Figure 3). Structures were built within the Cerius2 software [22] and geometry optimised at the HF/6-31G(d) level using Gaussian98 [23]. For each system two geometries were studied with different values of the O=C–O–R torsion angle φ. The lowest-energy conformation was found with φ = 0° (denoted cis) while the high-energy conformer has φ = 180° (denoted trans). Firstly, it was noted that the energy differences between the cis and trans conformers for 3a and 4a were identical (8.52 kcal mol–1). In contrast, the corresponding hydrolysis product in which either the citronellol or l-menthol components have been removed to afford the corresponding carboxylic acid, the energy difference between conformers was 6.24 kcal mol–1. The rotational energy profile of φ is similar for all three structures – the alcohol leaving group does not significantly affect the energy of the different conformers of a given molecule, thus steric repulsions between the alcohols and the remainder of the molecule are small. Plots of the electron density for the cis conformers of 3a and 4a (Figure 3) reveal that there is minimal difference in the local density around the carboxy group. While the isopropyl substituent of the cyclohexane ring in 4a-cis would appear to occlude the reactive carbonyl centre, the approach angle onto the C=O group will be perpendicular to the C=O vector and thus not close to the bulky ester group. Based upon this preliminary evidence, it appears that the observed lack of hydrolysis of 4 cannot not attributed to a predominant steric effect, and is most likely related to the poor leaving ability of the alcohol functionality. Indeed, one calculated difference between 3a and 4a are the partial atomic charges of the carbon atom of the COOR group, with values of –0.38 and –0.34 e, respectively. While this difference is only a small distinction, it is likely that the cause is the increased positive inductive effect offered by the leaving group in 4a cf. that of 3a, which promotes an increased negative (more positive) charge on the adjacent atoms.
Figure 3.
Plots of the total electron density of the cis conformers of 3a and 4a, drawn using the Molekel [24] software.
Figure 3.
Plots of the total electron density of the cis conformers of 3a and 4a, drawn using the Molekel [24] software.
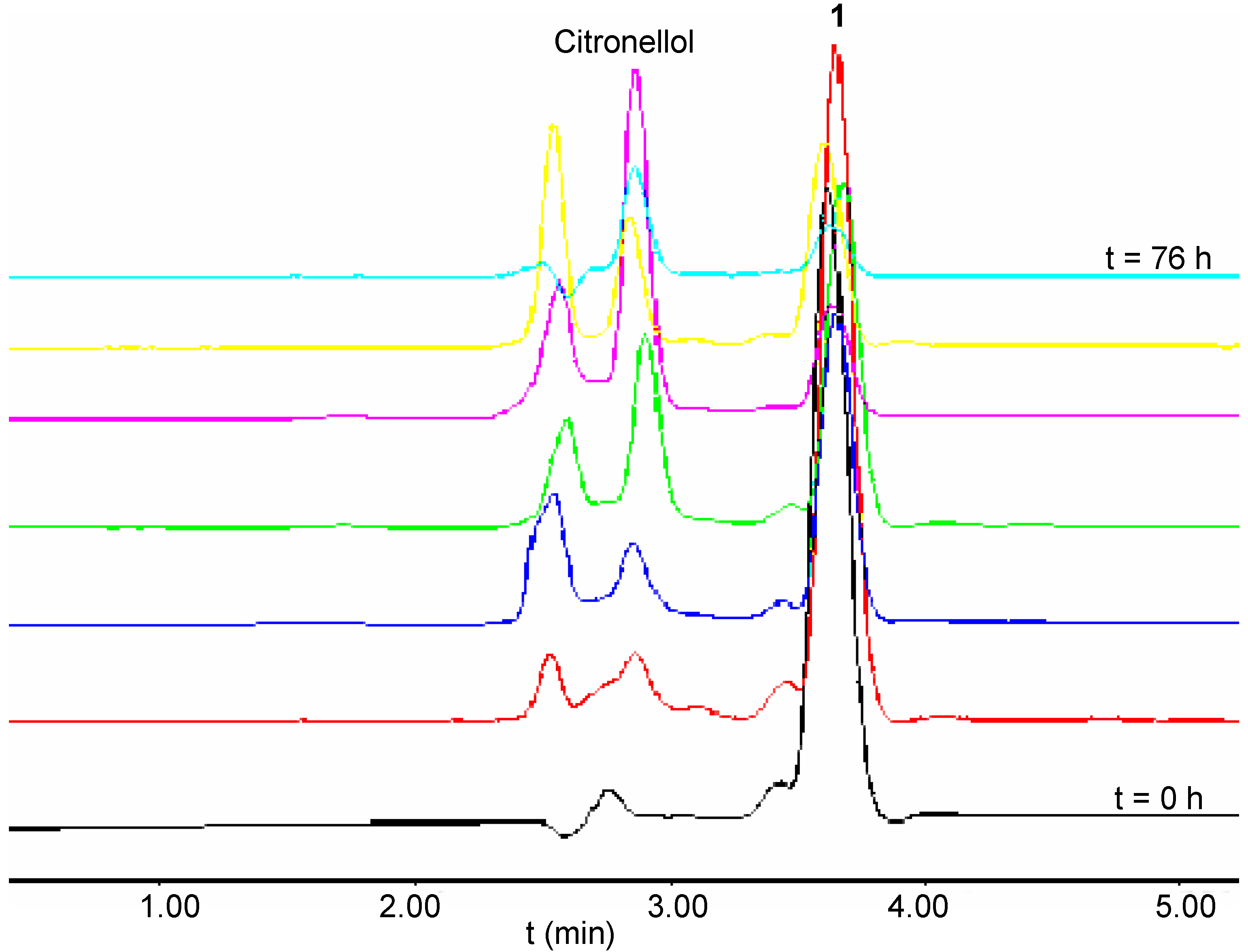
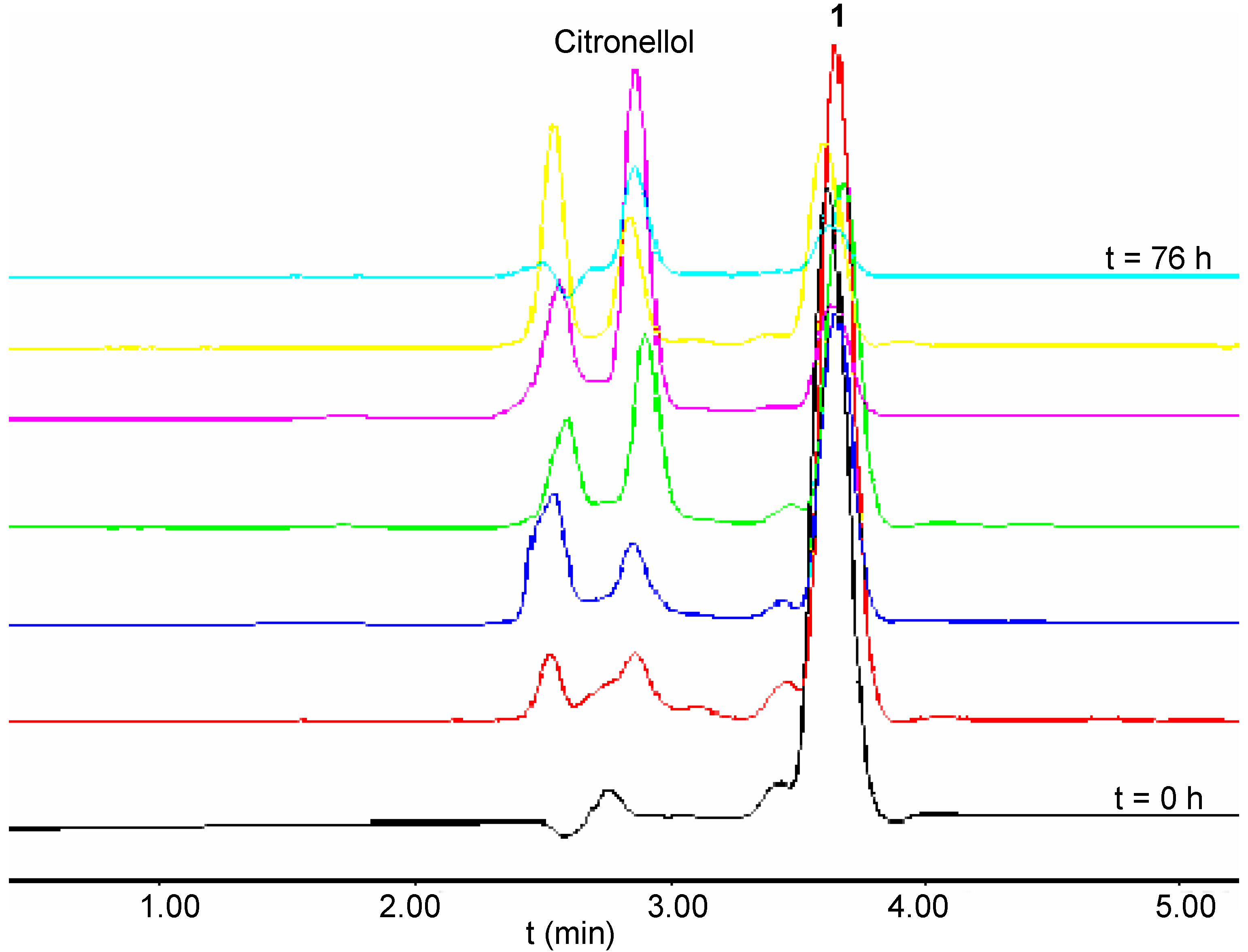
A study of the ester cleavage from the first generation polyamide dendrimers 5-8 was conducted by GC analysis. Although sample preparation was carried out under similar conditions used for the branched molecules 1-4, the chloroform solution resultant from the extraction of the aqueous mother liquor was injected directly into the GC column. This procedure eliminated one step in the sample preparation and decreased any potential error associated with this process. Furthermore, the analyses employed an internal standard to minimise the errors related with the sampling and injection processes. However, under the same hydrolytic conditions used in the other cases examined, release of the citronellol from C6-[G-1]3-N (7) was not observed. This result was rationalised as a consequence of two major factors: increased bulkiness of the dendritic structure which may have inhibited the access of the molecule into the enzyme active site, and also increased rigidity of the system introduced by the presence of the cinnamate moieties. Furthermore, it was speculated that the increased rigidity might also render the deprotonation process of the amide unit more difficult to achieve and consequently reduce the ability of intramolecular nucleophilic addition/elimination that was observed in the case of C3-[G-0]3-N (3) under mild basic conditions. Since the lipase from Candida cylindracia was found to catalyse the cleavage of the ester moieties only from C3-[G-0]3-N 3, it was decided to investigate another enzyme which belonged to the class of hydrolytic enzymes referred to as cutinases. The enzyme used was a solid-supported cutinase from Fusarium solani pisii, used commonly in detergent products. The release profiles for the series of citronellol-based dendrimers obtained under the optimum conditions for this enzyme, were very surprising. For example, C2-[G-0]2-C 1 underwent cleavage of the ester moieties (~80%) within the first 40 hours of hydrolysis. However, a decrease in concentration of citronellol was observed after this period of time, thereby indicating the occurrence of possible side reactions involving the alcohol (Figure 4).
Figure 4.
HPLC Traces of the Hydrolysis of 1 in the presence of a Cutinase, recorded at Different Times During the Reactions.
Figure 4.
HPLC Traces of the Hydrolysis of 1 in the presence of a Cutinase, recorded at Different Times During the Reactions.
In contrast, when the hydrolysis was carried out in the absence of the enzyme, significant release of alcohol was not detected. MALDI-TOF mass spectrometry did not reveal significant information regarding the hydrolytic pathway, although a decrease in intensity of the peaks corresponding to the starting material was observed with the proceeding of the reaction. Unfortunately, citronellol was not detectable under the conditions used in the mass spectrometric analysis, and also was not observed in the spectra of standard samples. Furthermore, analysis of negative ions via MALDI-TOF MS was not conducted, therefore, possible negatively charged species resulted from the hydrolysis were not observed. From the results obtained for the hydrolysis of 1, it was concluded that the hydrolysis of the ester moieties was enzyme-catalysed although it occurred without formation of the cyclic imide. The presence of the enzyme also appeared to influence much more significantly the hydrolytic process in comparison to the other enzyme tested (lipase) thus indicating more favourable interactions between the substrate and the enzyme active site. Similar behaviour was also observed in the case of the citronellol derived first generation polyamide dendrimers 5 and 7.
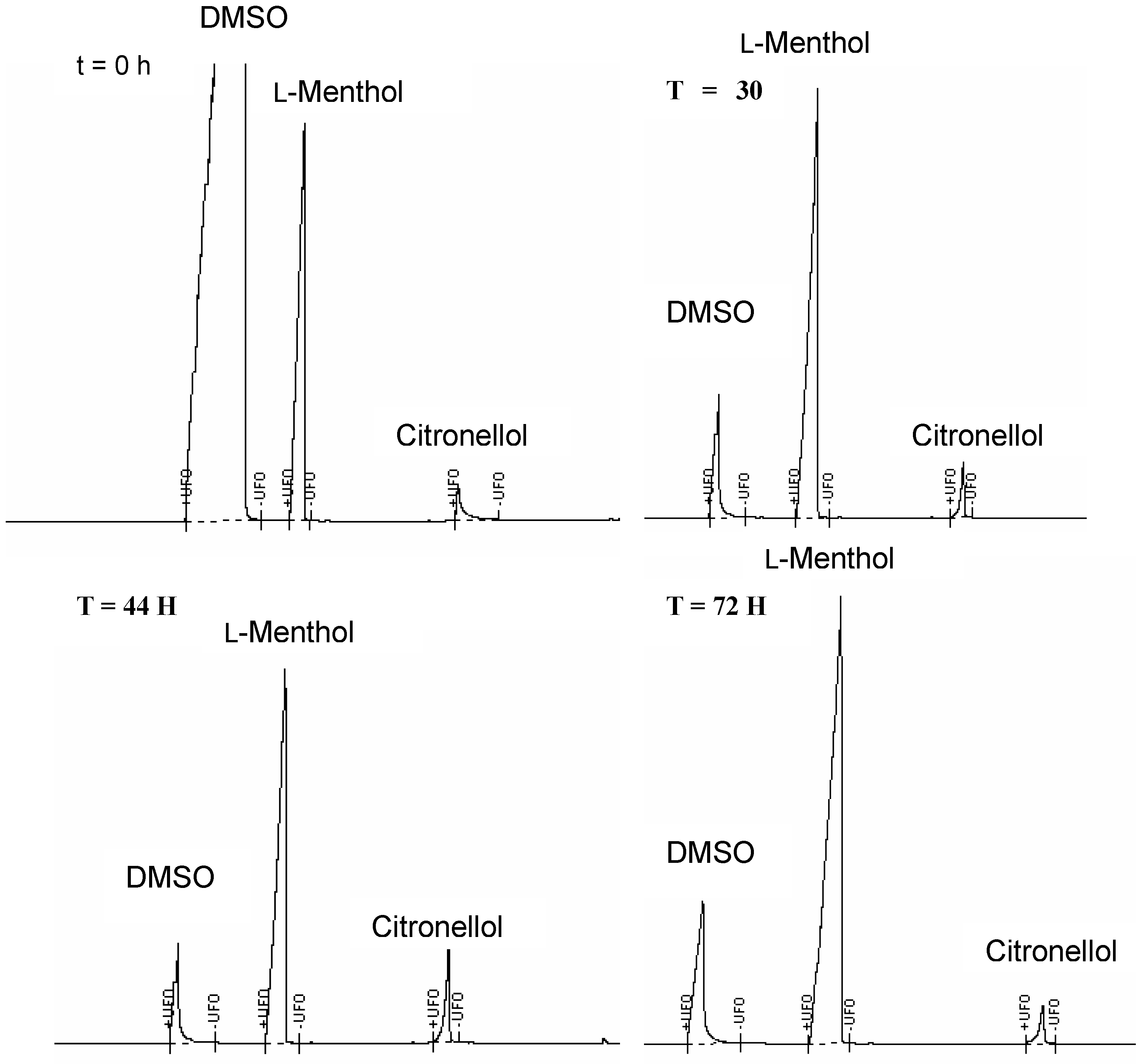
Figure 5.
GC Chromatograms of the Hydrolysis of C4-[G-1]2-C (5) in the Presence of Cutinase, Recorded after 0, 30, 44 and 72 Hours.
Figure 5.
GC Chromatograms of the Hydrolysis of C4-[G-1]2-C (5) in the Presence of Cutinase, Recorded after 0, 30, 44 and 72 Hours.
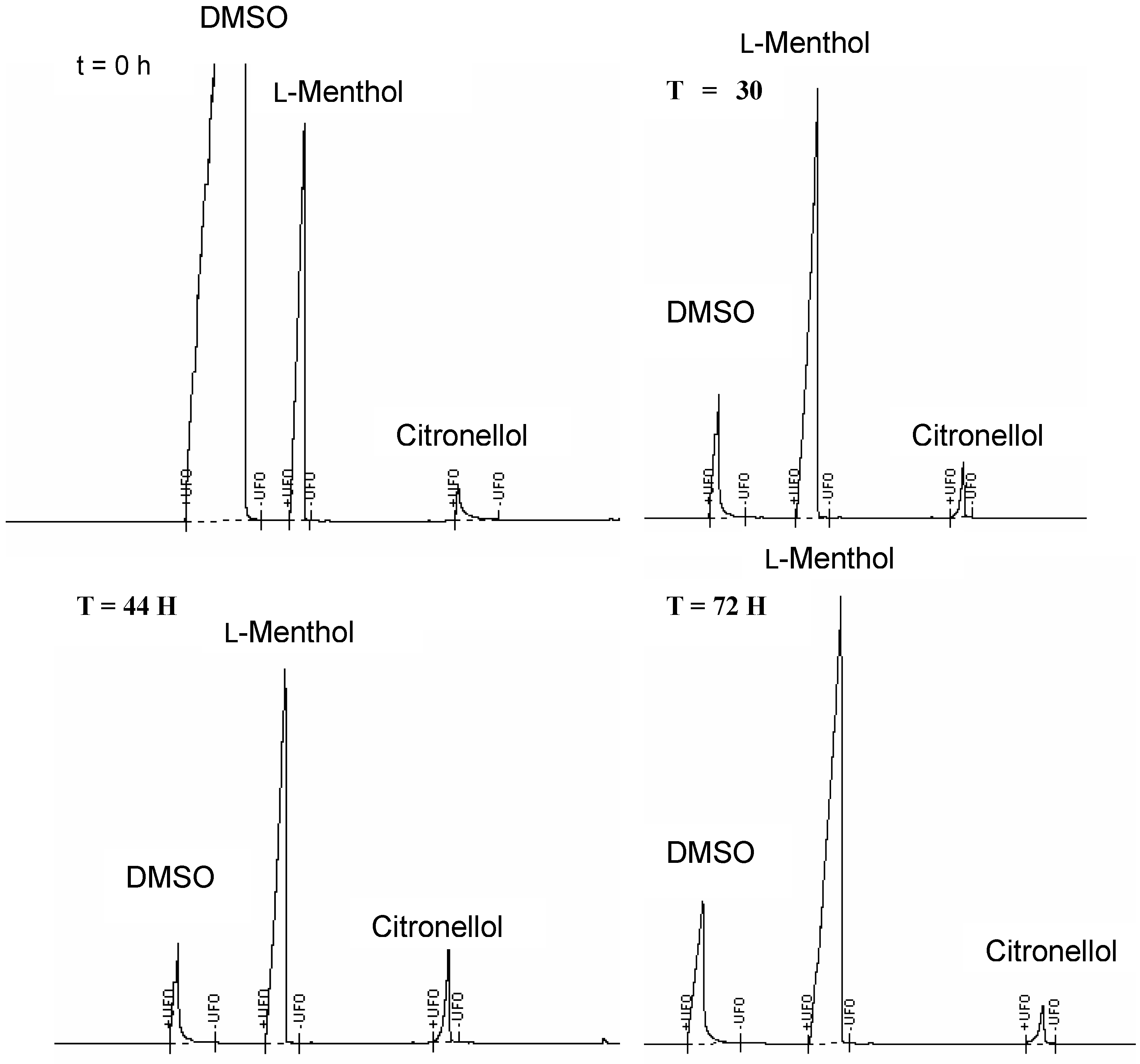
The analysis of the hydrolytic products was carried out using GC, in which each sample was spiked with l-menthol as the internal standard. When the hydrolysis was performed in the presence of a lipase, cleavage of the ester groups was not detected. However, improved results were obtained in the presence of the cutinase, whereas a small percentage of citronellol was released from the dendritic structures (~15-25%). The dendrimer C4-[G-1]2-C (5) exhibited release of citronellol over a period of 40 hours after which decrease in the concentration of citronellol was observed in a similar fashion as described above (Figure 5). Furthermore, the hydrolysis carried out in the absence of enzyme did not exhibit relevant release of citronellol, thus indicating that the ester cleavage was promoted by the enzyme rather than dictated by the reaction conditions. MALDI-TOF mass spectrometric analysis in this case also did not prove to be informative: – the almost complete absence of the peak corresponding to the dendritic starting material was observed from the beginning of the hydrolysis. This result was attributed to the experimental procedure required for the preparation of the analytical samples. Since the enzyme was solid supported, each sample was subjected to filtration and extraction with an organic solvent prior to analysis. However, the lack of the signal corresponding to the dendritic species in the MALDI-TOF mass spectrometric analysis led to the hypothesis that the dendrimers were absorbed onto the solid support and not eluted in the organic solvent used in the extraction step.
The polyamide dendrimer C6-[G-1]3-N (7) also exhibited similar behaviour in the presence of the cutinase, although the increase in concentration of citronellol was very moderate (~15%). This moderate hydrolytic process – as in the case of the hydrolysis carried out in the presence of a lipase – may be attributable to the increased rigidity of the structure C6-[G-1]3-N (7) when compared with C4-[G-1]2-C (5). The dendrimer C6-[G-1]3-N (7) was characterised by a tetrafunctional core with three arms possessing only two carbon chains. In contrast, C4-[G-1]2-C (5) possessed a bifunctional core with longer alkyl chains (seven methylenic residues), which imparted a higher degree of flexibility to the structure. Furthermore, 7 was characterised by the presence of three rigid polyamide dendrons around the core unit, with considerable increase of the bulkiness of the molecules.
The series of dendrimers bearing l-menthol esters at the periphery were also subjected to the hydrolytic conditions in the presence of the cutinase. However, as in the case of the hydrolysis in the presence of lipase, these molecules did not exhibit any evidence of alcohol release, confirming the selectivity of these classes of hydrolases towards esters of primary alcohols.
Conclusions
Following the pioneering work of Seebach and co-workers [9], the post-synthetic modification of dendritic surfaces was investigated by mean of two hydrolytic enzymes, a lipase and a cutinase. From the investigations carried out with the aid of analytical techniques such as RP-HPLC, GC and MALDI-TOF mass spectroscopy emerged the ability of cutinase to efficiently interact with the linear polyamides 1 and hence catalyse the alcohol release (up to 80% release). However, increasing the degree of branching and reducing the flexibility of the polyamides resulted in a significant reduction in the catalytic activity, probably dictated by the steric hindrance of these large branched molecules.
The lipase proved to be a less efficient catalyst for these branched substrates, since cleavage of the ester was only observed in the case of the polyamide C3-[G-0]3-N (3). However, this polyamide exhibited ester cleavage in a smaller extent also in the absence of the enzyme, via a neighbouring group participation effect, whereby the alcohol was released as a consequence of an intramolecular nucleophilic addition/elimination reaction. This process has been described in the literature [18], and occurs even under mild basic conditions. In this specific case, it was suggested that the tertiary amine in the core unit of the molecule acted as a base catalysing the deprotonation of the amide and hence the formation of the cyclic imide. The enzymatic ester cleavage of l-menthol esters was unsuccessful in all of the cases examined, thus indicating the selectivity of these hydrolytic enzymes towards esters derived from primary alcohols under these conditions.
Experimental
General
Reagents were purchased from the Aldrich Chemical Company, Acros Organics and Lancaster and were used as received without purification. Solvents were used as supplied, with the exception of the following: – tetrahydrofuran (THF) was distilled under nitrogen from sodium and benzophenone, dichloromethane (CH2Cl2) was distilled under nitrogen from calcium hydride. Solvents of analytical and HPLC-grade were purchased from Fisher Scientific and used as received without purification. Lipase from Candida cylindracea (943 unitmg-1) was purchased from Aldrich and used without purification, while cutinase (1% active enzyme protein) from Fusarium solani pisii was supplied by Unilever Research and Development, Port Sunlight, UK. Succinic acid mono-(5-isopropyl-2-methyl-cyclohexyl) ester (9) was prepared according to the method reported by Jabloner and Dunbar [13b]. The synthesis of the polyamide compounds described in this paper has been reported elsewhere [12]. MALDI-TOF mass spectra were obtained on a SAI LT3 LaserTof MALDI-TOF mass spectrometer using α-cyano-4-hydroxycinnamic acid as the matrix. A typical sample preparation is described as follows: 3 μL of a solution of the analyte in THF (1-10 mgmL-1) was combined with 10-20 μL of the freshly prepared matrix (0.1 or 0.2 M in THF) in a mini-vial, and from the mixture was taken a 2 μL aliquot which was transferred carefully onto a sample plate and left to air dry prior to analysis. Reverse-phase high performance liquid chromatographic analyses (RP-HPLC) were carried out using a Perkin-Elmer Series 410 LC pump in conjunction with a refractive index detector Polymer Laboratories Pl-RI 800 and interfaced with PL Caliber® LC/GC version 4 software (Polymer Laboratories, Ltd). Each sample (20 μL) was dissolved in CH3CN and injected (20 μL) onto a Phenonomex Luna C18 (2), 100 × 4.6 mm column (3 μm beads) equipped with a pre-column Hichrom C18 (5 μm), and temperature controlled at 40 °C. The flow rate of the mobile phase (CH3CN) was 1 mLmin-1. Gas chromatographic (GC) analyses were carried out using a Perkin-Elmer Autosystem XL gas chromatograph in conjunction with a flame ionisation detector (FID) and interfaced with TotalChrom™ workstation chromatography software (Perkin-Elmer Instruments). Each sample (dissolved in CHCl3) was injected (10 μL) onto a Supelcowax 10 column, 30 × 0.25 mm ID, 0.25 μm film, using helium as gas carrier at a pressure of 16 PSI. The temperature of the column was held at 50 °C for the first two minutes, then was increased to 180 °C with a ramp of 2.5 °Cmin-1 and finally held at 180 °C for 5 minutes.
Phosphate Buffer Preparation
0.01 M Phosphate buffer, pH 7.2 (100.00 mL) was prepared by combining 0.20 M sodium phosphate monobasic solution (8.40 mL) – (stock solution A), – 0.20 M sodium phosphate dibasic solution (21.60 mL) – (stock solution B) – and distilled water (60.00 mL). Stock solution A was obtained by dissolving Na2HPO4 (13.90 g, 0.10 mol) in distilled water (500.00 mL). The stock solution B was obtained dissolving NaH2PO4·12H2O (71.63 g, 0.20 mol) in distilled water (1000.00 mL). 0.10 M Phosphate buffer, pH 7.8 (200.00 mL) was prepared by combining 0.2 M sodium phosphate monobasic solution (8.50 mL) – (stock solution A), – 0.2 M sodium phosphate dibasic solution (91.50 mL) – (stock solution B) – and distilled water (100.00 mL). Stock solution A was obtained dissolving Na2HPO4 (2.40 g, 0.02 mol) in distilled water (100.00 mL). The stock solution B was obtained dissolving NaH2PO4·12H2O (7.16 g, 0.02 mol) in distilled water (100.00 mL).
Citronellol standards for RP-HPLC analyses:
Known aliquots of citronellol were weighed and transferred into clean, dry volumetric flasks (25 mL). CH3CN was added to volume in order to obtain final concentrations of 0.1, 0.5, 1.0, 2.0, 5.0 and 10.0 mgmL-1. The peak areas obtained from the RP-HPLC analyses were then plotted against the concentration in order to obtain a linear correlation between the two variables. The optimum correlation was obtained by using Equation 1:
where
Equation 1. Linear Correlation Between Peak Areas and Concentrations.
y = 0.0498X (R2 = 0.9207),
y = 0.0498X (R2 = 0.9207),
- × is the peak area
- y is the concentration.
- Using this equation the unknown concentration of samples of citronellol were determined.
Citronellol standards for GC analyses:
Known aliquots of citronellol were weighed and transferred into clean, dry volumetric flasks (25 mL). A solution of l-menthol in CHCl3 (10.0 mgmL-1) was added to obtain a final concentration of 1.0 mgmL-1. CHCl3 was then added to the volumetric flask up to the required volume in order to obtain final concentrations of 0.01, 0.05, 0.10, 0.50, 1.00 and 2.00 mgmL-1. The peak areas obtained from the GC traces were then plotted against the concentration and the correlation between the two variables was calculated according to the method of the internal normalisation, where the detector response factor (DRF value)25 is given by Equation 2:
where:
Equation 2. The Detector Response Factor.
- AC is the peak area of the citronellol;
- AM is the peak area of the internal standard, L-menthol;
- CM is the concentration of the internal standard, L-menthol and
- CC is the concentration of citronellol.
- Using this equation the unknown concentration of samples of citronellol were determined.
General Procedure for the Hydrolysis Studies in 0.01 mM Phosphate Buffer, pH= 7.2 at 37 °C in the Presence of Lipase from Candida Cylindracea: C2-[G-0]2-C (1).
The hydrolysis was conducted in a carousel reactor holding 12 sealed vials equipped with a temperature-controlling probe set at 37 °C. Into each vial was placed a solution of the enzyme (4.90 mL). The enzyme solution was prepared as follows: 24.0 mg of lipase from Candida cylindracea were transferred carefully into a 100 mL volumetric flask and the phosphate buffer was added up to the desired volume. The resultant suspension was subjected to ultrasound for 10 minutes prior to use. The enzyme suspension was left equilibrating at 37 °C for 2 hours prior the addition of a solution of C2-[G-0]2-C (1) in CH3CN (0.10 mL), obtained by dissolving 0.13 g of 1 in CH3CN (1.30 mL). After set periods of time the content of each vial was extracted with CHCl3 (2 × 5.00 mL). The solvent was then removed under reduced pressure, the residue dissolved in CH3CN (0.50 mL) and the solution analysed by RP-HPLC chromatography and MALDI-TOF mass spectrometry.
Hydrolysis studies in 0. 1 mM phosphate buffer, pH= 7.8 at 37 °C in the presence of Cutinase from Fusarium Solani pisii – typical procedure: C2-[G-0]2-C (1).
Into each vial were placed: the enzyme (1% active enzyme protein) (0.10 g) and the phosphate buffer (2.45 mL). The vials were then kept at 37 °C for a period of 2 hours prior to addition of a solution of C2-[G-0]2-C (1) in CH3CN (50 μL), obtained by dissolving 1 (0.10 g) in CH3CN (1.00 mL). After set intervals of time the content of each vial was filtered on a sintered funnel, washed with CHCl3 (6.00 mL) and then with distilled water (3.00 mL). The organic phase was separated (2.50 mL) and the solvent removed under reduced pressure. The residue was dissolved in CH3CN (0.30 mL) and analysed by RP-HPLC chromatography, GC chromatography and MALDI-TOF mass spectrometry.
Note: The hydrolysis studies of compounds M2-[G-0]2-C (2), C4-[G-1]2-C (5), M4-[G-1]2-C (6) and C6-[G-1]3-N (7) were carried according to the procedures described above. ‘Blank’ hydrolysis assays were carried out in exactly the same manner but in these cases the phosphate buffer added to each vial did not contain the enzyme.
Acknowledgements
F.A. would like to thank Unilever Research and Development for a postgraduate studentship. The authors would like to acknowledge EPSRC (GR/M91884) and SAI Ltd for the funding provided for the MALDI-TOF MS facilities at the University of Reading. W.H. would like to thank EPSRC (GR/M42824) for providing funds for the HPLC equipment used in this study.
References
- Koeller, K.M.; Wong, C.-H. Nature 2001, 409, 232.
- Carrea, G.; Riva, S. Angew. Chem., Int. Ed. Engl. 2000, 39, 2226.
- Klibanov, A.M. Nature 2001, 409, 241.
- Roberts, S.M. J. Chem. Soc., Perkin. Trans. 1 1999, 1.
- Kadereit, D.; Waldmann, H. Chem. Rev. 2001, 101, 3367.
- Reetz, M.T. Curr. Op. Chem. Biol. 2002, 6, 145. [CrossRef]
- Gross, R.A.; Kumar, A.; Kalra, B. Chem. Rev. 2001, 101, 2097.Kobayashi, S.; Uyama, H.; Kimura, S. Chem. Rev. 2001, 101, 3793.Kline, B.; Beckman, E.; Russell, A. J. Am. Chem. Soc. 1998, 120, 9475. [CrossRef]Al-Azemi, T.F.; Bisht, K.S. J. Polym. Sci. Part A: Polym. Chem. 2002, 40, 1267. [CrossRef]Bisht, K.S.; Deng, F.; Gross, R.A.; Kaplan, D.L.; Swift, G. J. Am. Chem. Soc. 1998, 120, 1363. [CrossRef]
- Hayashi, T.; Nakayama, K.; Mochizuki, M.; Masuda, T. Pure Appl. Chem. 2002, 74, 869.
- Seebach, D.; Herrmann, G.F.; Lengweiler, U.D.; Bachmann, B.M.; Amrein, W. Angew. Chem., Int. Ed. Engl. 1996, 35, 2795.
- Fréchet, J.M.J.; Hawker, C.J. Comprehensive Polymer Science 2nd suppl.; Aggarval, S.L., Russo, S., Eds.; Pergamon Press: New York, 1996; p. 160. [Google Scholar]
- Hawker, C. J.; Fréchet, J. M. J. J. Chem. Commun., Chem. Commun. 1990, 1010.
- (a)Aulenta, F.; Foster, A.; Rannard, S.; Thornthwaite, D.W.; Hayes, W. submitted.Aulenta, F. PhD Thesis, The University of Reading, Reading, U.K., 2003.
- Cotterill, A.S.; Gill, M.; Milanovic, N.M. J. Chem. Soc., Perkin Trans. 1 1995, 1215.Jabloner, H.; Dunbar, B.I. J. Polym Sci.: Polym. Chem. Ed. 1980, 18, 2933. [CrossRef]
- Bodanszky, M.; Bodanszky, A. The Practice of Peptide Synthesis, 2nd Ed. ed; Springer Verlag: Berlin, 1994. [Google Scholar]
- Chowdhury, A.R.; Tripathi, M.K.; Jinwal, U.K.; Roy, U.; Kumar, U.V.; Bhaduri, A.P. J. Chem. Res. 1999, (S), 266.Graffner-Nordberg, M.; Sjödin, K.; Tunek, A.; Hallberg, A. Chem. Pharm. Bull. 1998, 46, 591. [CrossRef]
- Werbin, H.; Spoerri, P.E. J. Am. Chem. Soc. 1947, 69, 1681. [CrossRef]Feuer, H.; Harmetz, R. J. Am. Chem. Soc. 1958, 80, 5877. [CrossRef]
- Matsumoto, H.; Hamawaki, T.; Ota, H.; Kimura, T.; Goto, T.; Sano, K.; Hayashi, Y.; Kiso, Y. Bioorg. Med. Chem. Lett. 2000, 10, 1227.Matsumoto, H.; Kimura, T.; Hamawaki, T.; Kumagai, A.; Goto, T.; Sano, K.; Hayashi, Y.; Kiso, Y. Bioorg. Med. Chem. Lett. 2001, 9, 1589. [CrossRef]
- Bodanszky, M.; Kwei, J.Z. Int. J. Peptide Protein Res. 1978, 12, 69. [CrossRef]
- Gotor, V.; Brieva, R.; Rebolledo, F. Tetrahedron Lett. 1988, 29, 6973.Brieva, R.; Rebolledo, F.; Gotor, V. Chem. Commun. 1990, 1386.
- Astorga, C.; Rebolledo, F.; Gotor, V. Synthesis 1993, 287.
- Frérot, E.; Herbal, K.; Hermann, A. Eur. J. Org. Chem. 2003, 967.
- CERIUS2,version 3.0; Accelerys Inc.: San Diego, California, USA, 2001.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, K.M.Q.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Ortiz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P.M.W.; Johnson, B.G.; Chen, W.; Wong, M.W.; Andres, J.L.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian 98, Revision A.9; Gaussian, Inc.: Pittsburg, PA, 1998. [Google Scholar]
- Flükiger, P.; Lüthi, H.P.; Portmann, S.; Weber, J. Molekel v4.3; Swiss Centre for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]
- Jones, R.A. An Introduction to Gas-Liquid Chromatography; Academic Press: London and New York, 1970. [Google Scholar]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Aulenta, F.; Drew, M.; Foster, A.; Hayes, W.; Rannard, S.; Thornthwaite, D.; Youngs, T. Fragrance Release from the Surface of Branched Poly (Amide) S. Molecules 2005, 10, 81-97. https://doi.org/10.3390/10010081
AMA Style
Aulenta F, Drew M, Foster A, Hayes W, Rannard S, Thornthwaite D, Youngs T. Fragrance Release from the Surface of Branched Poly (Amide) S. Molecules. 2005; 10(1):81-97. https://doi.org/10.3390/10010081
Chicago/Turabian StyleAulenta, F., M. Drew, A. Foster, W. Hayes, S. Rannard, D. Thornthwaite, and T. Youngs. 2005. "Fragrance Release from the Surface of Branched Poly (Amide) S" Molecules 10, no. 1: 81-97. https://doi.org/10.3390/10010081