Eco-friendly Oxidative Iodination of Various Arenes with Sodium Percarbonate as the Oxidant†

Chair and Laboratory of Organic Chemistry, Faculty of Pharmacy, Medical University, 1 Banacha Street, 02 – 097 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(10), 1307-1317; https://doi.org/10.3390/10101307
Submission received: 17 May 2005
/
Revised: 12 July 2005
/
Accepted: 13 July 2005
/
Published: 31 October 2005
Abstract
:Six easy laboratory procedures are presented for the oxidative iodination of various aromatics, mostly arenes, with either molecular iodine or potassium iodide (used as the sources of iodinating species, I+ or I3+), in the presence of sodium percarbonate (SPC), a stable, cheap, easy to handle, and eco-friendly commercial oxidant.
Introduction
Aromatic iodides are generally more reactive, albeit are more costly, than the respective bromides and chlorides. There are many different methods, direct and indirect, for their synthesis [1], and they are widely used in organic synthesis in chemical laboratories and, to a lesser extent, in industry. Moreover, they are able to form a variety of aromatic hypervalent iodine derivatives, which have found increasing applications in modern organic synthesis [2]. Our two latest reviews [3,4] relate and explain a variety of direct oxidative iodination methods, suitable for both activated and deactivated aromatics, mostly arenes, devised in our laboratory since 1990, as well as our novel methods for preparing several classes of aromatic hypervalent iodine compounds, easily attainable from aromatic iodides. See also our former paper [5].
Three solid commercial products, viz. a urea–hydrogen peroxide adduct (UHP), H2NCONH2· H2O2, sodium perborate (SPB), NaBO3·H2O or NaBO3·4H2O, and sodium percarbonate (SPC), 2 Na2CO3·3H2O2, may be considered as ‘dry carriers’ of the hazardous and unstable hydrogen peroxide, are easy to handle, safe and stable at room temperature. Their ability to release oxidative species in water and organic media has made them useful oxidants in organic synthesis [6,7]. The oxidative iodination reactions with using UHP [5] or SPB [7] were already reported. However, so far nobody has used SPC as a cheap [8] and eco-friendly oxidant in the oxidative iodination reactions of various aromatics, both activated and deactivated ones, which has been the aim of our present work (vide infra).
Results and discussion
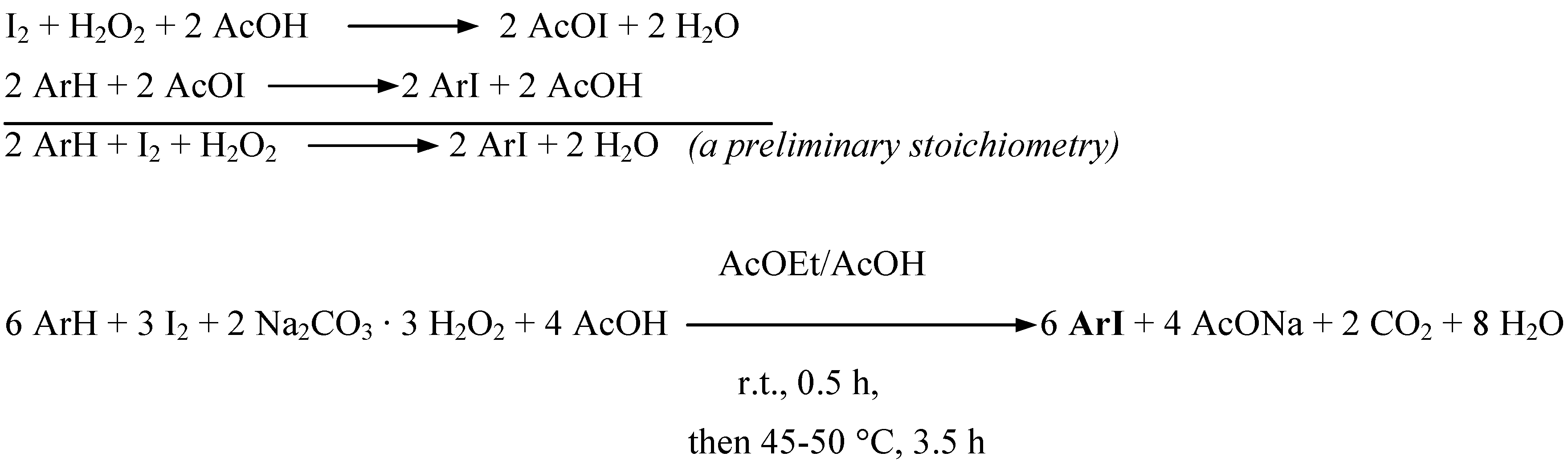
First we have attempted to oxidatively monoiodinate four exemplary aromatic amines (Table 1) according to the stoichiometry shown in Scheme 1 (Procedure 1).
Scheme 1.

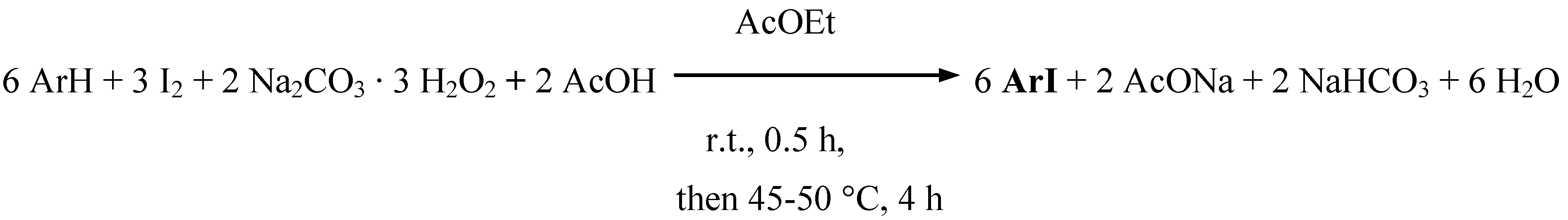
Supposedly, only some transient iodine(I) species, I+ i.e. AcOI, are preponderantly acting here as weak electrophiles. Powdered diiodine was suspended in a mixture made up of ethyl acetate and glacial acetic acid (the latter was used in a large excess to decompose in full all Na2CO3, releasing H2O2). SPC (in a 14% excess) was slowly added portionwise with stirring. Next, an aromatic amine was added, and the reaction mixture thus obtained was stirred first at room temperature for 30 min, and next at 45−50 ºC for ca. 3.5 hours. The reactions were complete when the iodine coloration faded. After cooling, the reaction mixtures were quenched by pouring into excess aqueous Na2SO3 solutions (a reductant, used to destroy unreacted diiodine and any oxidized species). The oily or solid crude products were typically isolated and purified (see the Experimental section for details) to give the purified iodinated products in 67−86% yields (Table 1). When aniline was diiodinated in 85% yield, we added only half of the amount, i.e. 3 H-Ar-H equivalents (Scheme 1), to the starting iodinating mixture. However, N,N-dimethylaniline and 2-chloroaniline gave only some tarry products under the above reaction conditions. Such easily oxidizable aromatic amines were however readily monoiodinated according to the stoichiometry shown in Scheme 2 (Procedure 2).
Scheme 2.

By comparing Scheme 1 and Scheme 2 one may deduce that the reaction media in Procedure 2 were notably less acidic, which was favorable for the successful oxidative iodination of the two aromatic amines. With the exception of this single important difference, the remaining reaction conditions and the reaction mechanism in Procedure 2 were nearly the same as those in Procedure 1, giving 4-iodo-N,N-dimethylaniline and 2-chloro-4-iodoaniline in 60 and 73% (purified) yields, respectively. However, the comparative monoiodinations of 2-bromoaniline and 2-toluidine were less effective than those in Procedure 1 (Table 1).
Uracil, benzene, two halobenzenes, and five weakly deactivated arenes (Table 1) were effectively oxidatively monoiodinated according to the stoichiometry shown in Scheme 3 (Procedure 3).
Scheme 3.
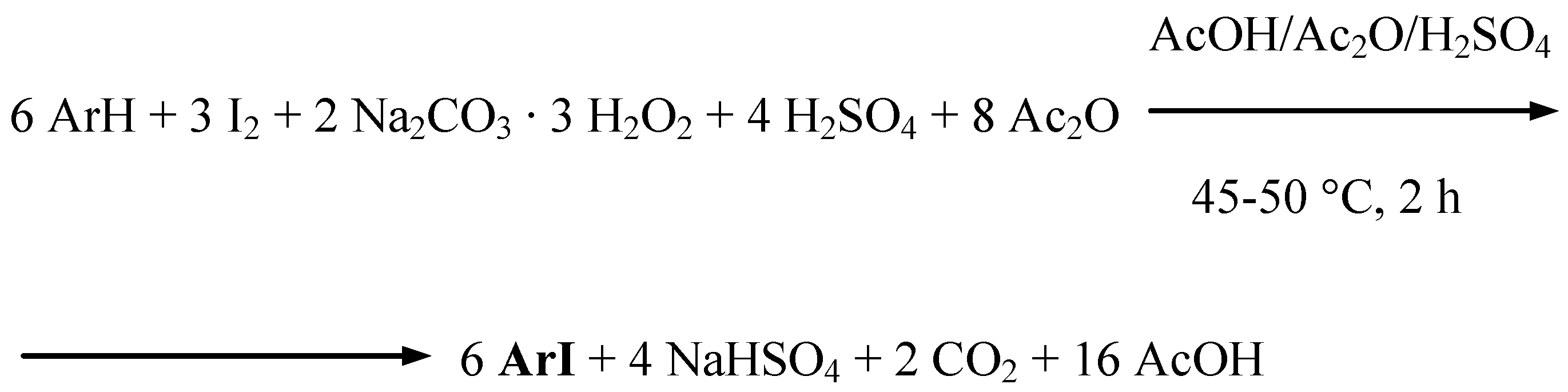
Supposedly, only some transient iodine(I) species, I+, i.e. IOSO3H, are preponderantly acting there as moderate electrophiles. As above, powdered diiodine and SPC (in a 54% excess), and next a chosen arene were suspended in anhydrous AcOH/Ac2O mixtures, then the mixtures were strongly acidified with varied amounts [9] of conc. (98%) H2SO4 slowly added dropwise with stirring and keeping the temperature below 10 ºC. The reaction mixtures were next heated and stirred for 2 hours at 45−50 ºC, where the following iodinating reactions underwent: ArH + IOSO3H → ArI + H2SO4. The reactions were quenched as above by pouring the final reaction mixtures into excess aqueous Na2SO3 solutions. The crude products were collected by filtration and worked up typically to give the purified iodinated products in 40−92% yields. When benzene was diiodinated in 83% yield, only half of the amount, i.e. 3 H-Ar-H equivalents (Scheme 3), was added to the starting iodinating mixture.
Five more strongly deactivated arenes were oxidatively monoiodinated according to the stoichiometry shown in Scheme 4 (Procedure 4).
Scheme 4.
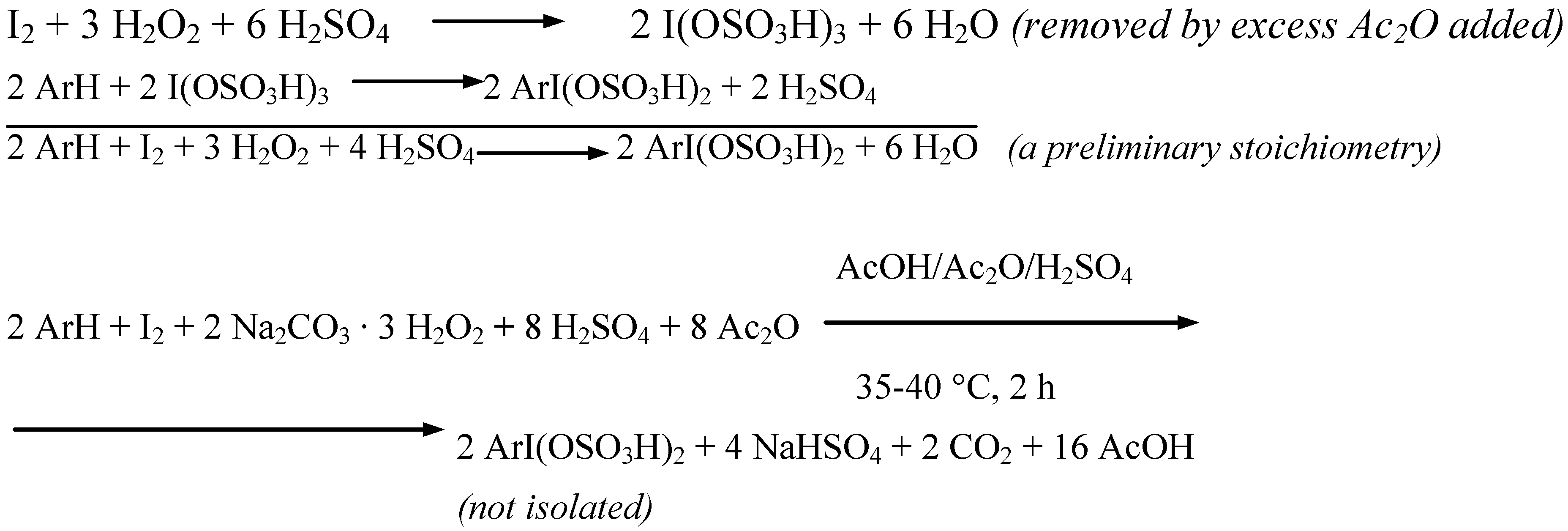
The oxidative iodination reactions were carried out in anhydrous AcOH/Ac2O mixtures, containing diiodine, the said arenes and SPC, and next they were strongly acidified with varied amounts [9] of conc. (98%) H2SO4 (a catalyst and reactant, which also decomposed in full all Na2CO3, with releasing H2O2), while keeping the temperature below 10 ºC. SPC was applied here in a 53% excess in respect to the amount demanded by the stoichiometry shown in Scheme 4. But in the said reaction mixtures some strongly electrophilic iodinating intermediates, I3+ i.e. I(OSO3H)3 [10], were generated, which readily reacted with the arenes to form the assumed, soluble organic iodine(III) intermediates, ArI(OSO3H)3 [10]. After completing the reactions carried out for 2 hours at 35−40 ºC, the cooled final reaction mixtures were poured into excess aqueous Na2SO3 solutions to destroy any oxidizing and oxidized species and unreacted diiodine; see Scheme 5.
Scheme 5.

The crude monoiodinated products were isolated and purified similarly to those obtained in Procedures 1−3 (see the experimental section for details) to give pure monoiodinated arenes in 60−94% yields. When benzene and benzophenone were oxidatively diiodinated, we added only half of the amount, i.e. 1 H-Ar-H equivalent (Scheme 4), to the starting reaction mixtures; the purified 1,4-diiodobenzene was obtained in 83% yield, while pure 3,3’-diiodobenzophenone was afforded in 51% yield (Table 1). However, nitrobenzene was likewise monoiodinated in only ca. 5% yield; cf. Ref. 5.
Anisole, two methoxynaphthalenes, and (for comparison) aniline were oxidatively monoiodinated using potassium iodide as the source of iodine(I) transient species, IOSO3H. According to Merkushev [1c], a possibility of replacement of elemental iodine, usually requiring its careful grinding before use, by readily accessible and cheap alkali iodides, is often convenient − albeit larger quantities of the oxidants are spent in such oxidative iodination reactions. The said four monoiodination reactions obeyed the stoichiometry shown in Scheme 6 (Procedure 5).
Scheme 6.
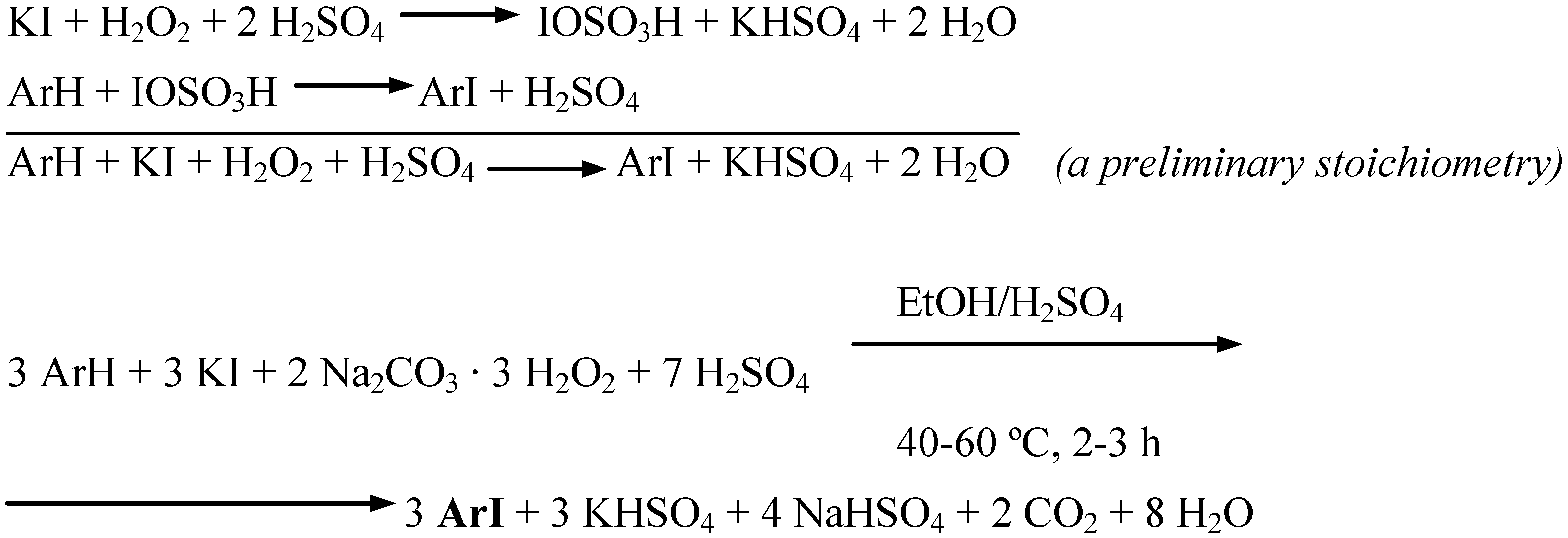
Potassium iodide dissolved in a little water was added to stirred ethanol (which can be replaced by methanol). A chosen arene and next SPC (used in ca. 54% excess) were added portionwise with stirring. Next, a definite volume of conc. H2SO4 was slowly added dropwise with stirring, while keeping the temperature below 10 ºC. The reaction mixtures thus obtained were stirred and heated under a reflux condenser for 2−3 hours at 40−60 ºC; for more details see the experimental section. After cooling, the final reaction mixtures were poured into vigorously stirred CH2Cl2/2% aq. Na2SO3 biphasic mixtures. The separated organic layers dried over Na2SO4 were filtered, next the solvent was distilled off. The solid residues were recrystallized to give pure monoiodinated products in 53−68% yields. Recently, Iskra and co-workers [9] oxidatively iodinated several highly activated arenes with one equivalent of KI and two equivalents of 30% aqueous hydrogen peroxide solution, in methanol in the presence of conc. H2SO4 − it is close to our Procedure 5, however with avoiding the use of 30% aqueous H2O2.
2- and 4-nitrophenol, 4-cresol, and 8-hydroxyquinoline were oxidatively diiodinated in 50% (v/v) aqueous acetic acid, using KI as the source of iodine(I) transient species, IOAc. The stoichiometry shown in Scheme 7 was obeyed (Procedure 6).
Scheme 7.
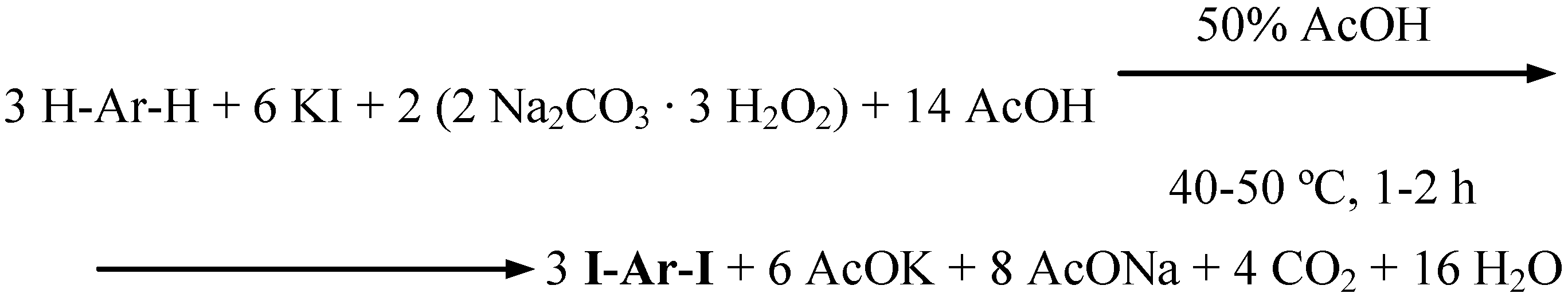
A definite amount of KI, a chosen phenol, and next SPC (used in ca. 84% excess) were subsequently slowly added to stirred 50% (v/v) aqueous acetic acid. The stirring was continued for 1−2 hours at 40−50 ºC. The cooled final reaction mixtures were poured into vigorously stirred CH2Cl2/2% aq. Na2SO3 biphasic mixtures. The separated organic layers dried over Na2SO4 were filtered, the solvent was distilled off, and the residues were recrystallized to give pure diiodinated products in 59−85% yields (Table 1). For comparison, we likewise monoiodinated aniline and 4-iodoaniline to obtain pure 4-iodo- and 2,4-diiodoaniline in 77 and 59% yields, respectively, with using the stoichiometry shown in Scheme 7, but corrected as follows: let one inserts there 6 ArH in place of 3 H-Ar-H to obtain the respective 6 ArI equivalents of the monoiodinated products.
Conclusions
Summing up, our easy and widely varied iodinating Procedures 1−6 reported in this paper gave mono- or diiodinated products from a large variety of different aromatics: highly activated arylamines and phenols, benzene, uracil, halobenzenes, and some activated or deactivated arenes in moderate to good yields (Table 1). The novel application of an eco-friendly, cheap [8], stable and easy to handle oxidant, viz. sodium percarbonate (SPC), makes our iodination methods to be attractive for organic chemists. In our opinion, the presented herein iodination reactions can be safely scaled up.
Experimental
General
The melting points of the freshly purified iodinated products are uncorrected and are compared with the literature data (Table 1). Their homogeneities were checked by TLC, next they were microanalyzed (% I ± 0.4), and finally their 1H and 13C NMR spectra (not shown here) were recorded at r.t. with a Brucker Avance DMX 400 MHz spectrometer in appropriate solvents, next compared with the same spectra of authentic samples and/or computed theoretical spectra. All the reagents and solvents were commercial (Aldrich, Lancaster) and were used without further purification. Elemental iodine (diiodine) should be finely powdered to facilitate its dissolution in the reaction mixtures. SPC (Aldrich) used in our iodinating experiments contained ca. 25% H2O2 [8] (theoretically, for 2 Na2CO3∙ 3H2O2 calc. 32.5% H2O2).
Optimized iodinating procedures, with using sodium percarbonate (SPC) as the oxidant
Procedure 1, applicable for some arylamines
Powdered diiodine (0.51 g, 2.0 mmol; 0% excess) was suspended in a mixture made of AcOEt (8 mL) and glacial AcOH (10 mL), then SPC (0.31 g, 2.3 mmol H2O2; 14% excess) was slowly added portionwise, with stirring, within 20−30 min, next followed by an aromatic amine (4.2 mmol; 5% excess to prevent the possible diiodination) [when aniline was diiodinated, only 2.0 mmol (0% excess) of the aniline was added]. The stirring was continued for 30 min at r.t., next the temperature was raised to 45−50 ºC, and the stirring was continued for a further 3.5 h under a reflux condenser. After cooling, the reaction mixtures were slowly added to stirred aq. Na2SO3 solutions (1 g Na2SO3 dissolved in 70 mL water). The precipitated crude products were collected by filtration, washed well with cold water until the filtrates were neutral, dried preliminarily by the suction, and next air-dried in the dark; they were recrystallized from appropriate organic solvents. If the semisolid crude products could not be efficiently isolated, they were extracted with CHCl3 (3 x 10 mL), the combined extracts were washed with 2% aq. Na2SO3 and water, dried over anhydr. Na2SO4, filtered, the solvent was distilled off, and the solidified residues were recrystallized from appropriate organic solvents to give the purified iodinated products in the yields shown in Table 1.
Procedure 2, applicable for easily oxidizable arylamines
Powdered diiodine (0.51 g, 2.0 mmol; 0% excess) was suspended in a mixture made of AcOEt (20 mL) and glacial AcOH (0.11 mL, 2.0 mmol), and SPC (0.31 g, 2.3 mmol H2O2; 14% excess) was slowly added portionwise, with stirring, within 20−30 min, next followed by an aromatic amine (4.2 mmol; 5% excess to prevent the possible diiodination). The reaction mixture was stirred at r.t. for 30 min and next at 45−50 ºC for 4 h under a reflux condenser [only for the iodination of 2-toluidine: at 50−55 ºC for 6 h]. After cooling, it was slowly poured, with stirring, into an aq. Na2SO3 solution (1 g Na2SO3 dissolved in 70 mL water) and the crude solid product was collected by filtration. The following workups were the same as those in Procedure 1 to give pure iodinated products in the yields shown in Table 1.
Procedure 3, applicable for benzene and some weakly deactivated arenes
Powdered diiodine (0.51 g, 2.0 mmol; 0% excess) was suspended in a mixture made of glacial AcOH (6 mL) and Ac2O (3 mL), and next SPC (0.42 g, 3.1 mmol H2O2; 54% excess) was slowly added portionwise, with stirring, within 20−30 min. The stirred mixture was slowly warmed up to 30−35 ºC, and a chosen arene (4.2 mmol; 5% excess to diminish the possible diiodination) or benzene (2.0 mmol; 0% excess – for its diiodination) were added. After cooling the reaction mixtures to 5−10 ºC, the given below varied amounts [9] of conc. (98%) H2SO4 were slowly added dropwise with stirring and keeping the temperature below 10 ºC:
- (a)
- 4.26 mL H2SO4 (7.84 g; 80 mmol) for the iodination of C6H5NHAc or uracil;
- (b)
- 4.80 mL H2SO4 (8.83 g; 90 mmol) for the iodination of C6H6, C6H5Br or C6H5Cl;
- (c)
- 5.33 mL H2SO4 (9.80 g; 100 mmol) for the iodination of 4-MeC6H4COOH, 4-MeC6H4COOMe, 4-O2NC6H4Me or 4-O2NC6H4OMe;
- (d)
- 7.50 mL H2SO4 (13.8 g; 140 mmol) for the diiodination of PhH.
Procedure 4, applicable for benzene and some more strongly deactivated arenes
Powdered diiodine (0.56 g, 2.2 mmol; 10% excess) was suspended in a mixture made of glacial AcOH (8 mL) and Ac2O (5 mL), and next SPC (1.25 g, 9.2 mmol H2O2; 53% excess) was slowly added portionwise, with stirring, within 20−30 min. The reaction mixture was slowly warmed up to 30−35 ºC, and a deactivated arene (4.0 mmol; 0% excess) or benzene, or benzophenone (2.0 mmol; 0% excess – for the diiodination reactions) were added. After cooling the reaction mixtures to 5−10 ºC, the given below varied amounts [9] of conc. (98%) H2SO4 were slowly added dropwise with stirring and keeping the temperature below 10 ºC:
- (a)
- 3.60 mL H2SO4 (6.60 g; 67.5 mmol) for the iodination of PhCOOH;
- (b)
- 4.80 mL H2SO4 (8.83 g; 90.0 mmol) for the iodination of PhI, PhCOOMe, 4-MeC6H4COOH, 4-O2NC6H4Me, and for the diiodination of PhH;
- (c)
- 6.80 mL H2SO4 (12.5 g; 127 mmol) for the diiodination of PhCOPh.
Procedure 5, applicable for some aromatic ethers and aniline
KI (0.66 g, 4.0 mmol; 0% excess) prior dissolved in a little water (ca. 1 mL) was added with stirring to ethanol (20 mL). An aromatic ether or aniline (4.2 mmol; 5% excess to prevent the diiodination) and next SPC (0.84 g, 6.2 mmol H2O2; 54% excess) were slowly added portionwise with stirring, within 20−30 min. After cooling the mixtures to 5−10 °C, conc. (98%) H2SO4 (0.6 mL, 1.1 g; 11.2 mmol) was slowly added dropwise with stirring and keeping the temperature below 10 ºC. Next, the reaction mixtures were stirred and heated under a reflux condenser as follows:
- (a)
- for 2 h at 40 °C for the iodination of PhOMe;
- (b)
- for 2 h at 50−60 °C for the iodination of 1- or 2-MeOC10H7;
- (c)
- for 3 h at 40 °C for the iodination of PhNH2.
Procedure 6, applicable for some phenols and aromatic amines
(a) For the diiodination of four phenols, KI (1.4 g, 8.5 mmol; 6.1% excess) prior dissolved in a little water (ca. 2 mL) was added with stirring to 50% (v/v) aq. AcOH (30 mL) containing a given phenol (4.0 mmol; 0% excess), next followed by slow addition of SPC (2.0 g, 14.7 mmol H2O2; 84% excess) within 20−30 min. The stirring was continued for 2 h at 40−50 °C under a reflux condenser. After cooling, the final reaction mixtures were slowly poured into vigorously stirred biphasic mixtures made of CH2Cl2 (50 mL) and 2% aq. Na2SO3 (40 mL). The following workups were the same as those in Procedure 5 to obtain pure diiodinated phenols in the yields given in Table 1.
(b) For the monoiodination of PhNH2 or 4-IC6H4NH2, KI (1.3 g, 8.0 mmol; 0% excess) prior dissolved in a little water (ca. 2 mL) was added with stirring to a solution of an aromatic amine (8.4 mmol; 5% excess to prevent the diiodination) in 50% (v/v) aq. AcOH (30 mL). SPC (2.0 g, 14.7 mmol H2O2; 84% excess) was slowly added portionwise with stirring within 20−30 min. The stirring was continued for 1 h at 40−50 °C under a reflux condenser. After cooling, the final reaction mixtures were slowly poured into vigorously stirred biphasic mixtures made of CH2Cl2 (50 mL) and 2% aq. Na2SO3 (40 mL). The following workups were the same as those in Procedure 5 to obtain pure monoiodinated arylamines in the yields given in Table 1.
Note. The final yields given in Table 1 for the purified iodinated products were calculated from the amounts of those reagents (arenes, diiodine or KI) which were used in the oxidative iodination reactions in strictly stoichiometric quantities (0% excess).

{kind=link}
{kind=link}
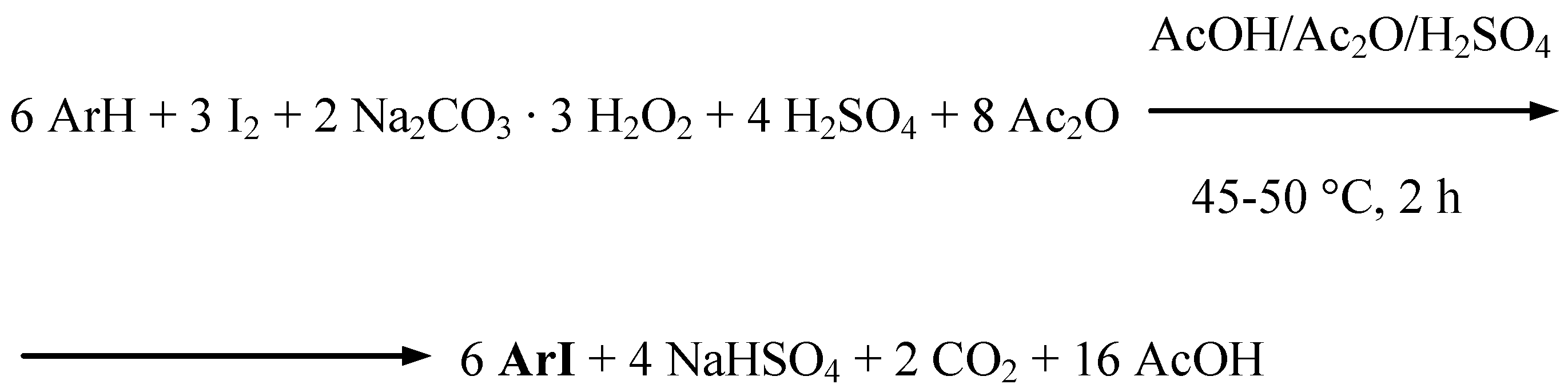{kind=link}
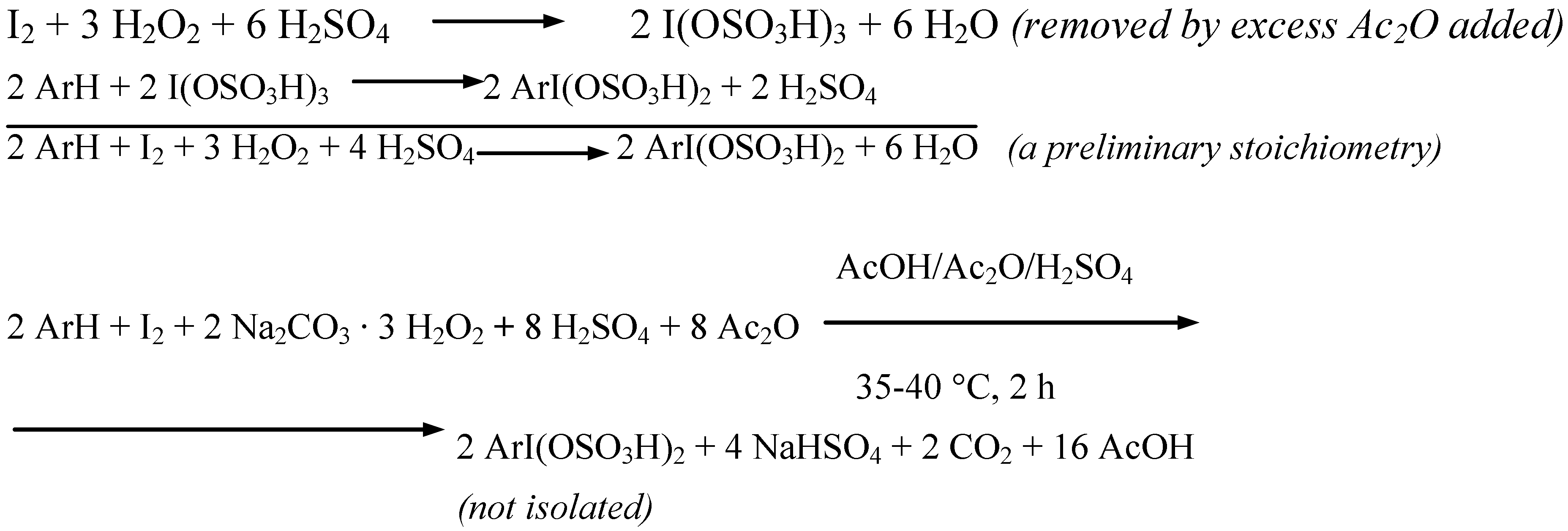{kind=link}
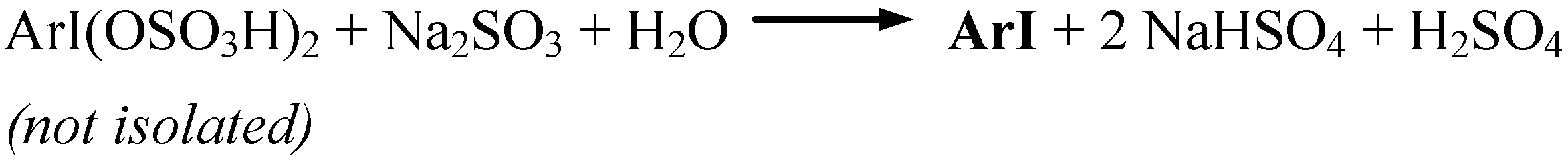{kind=link}
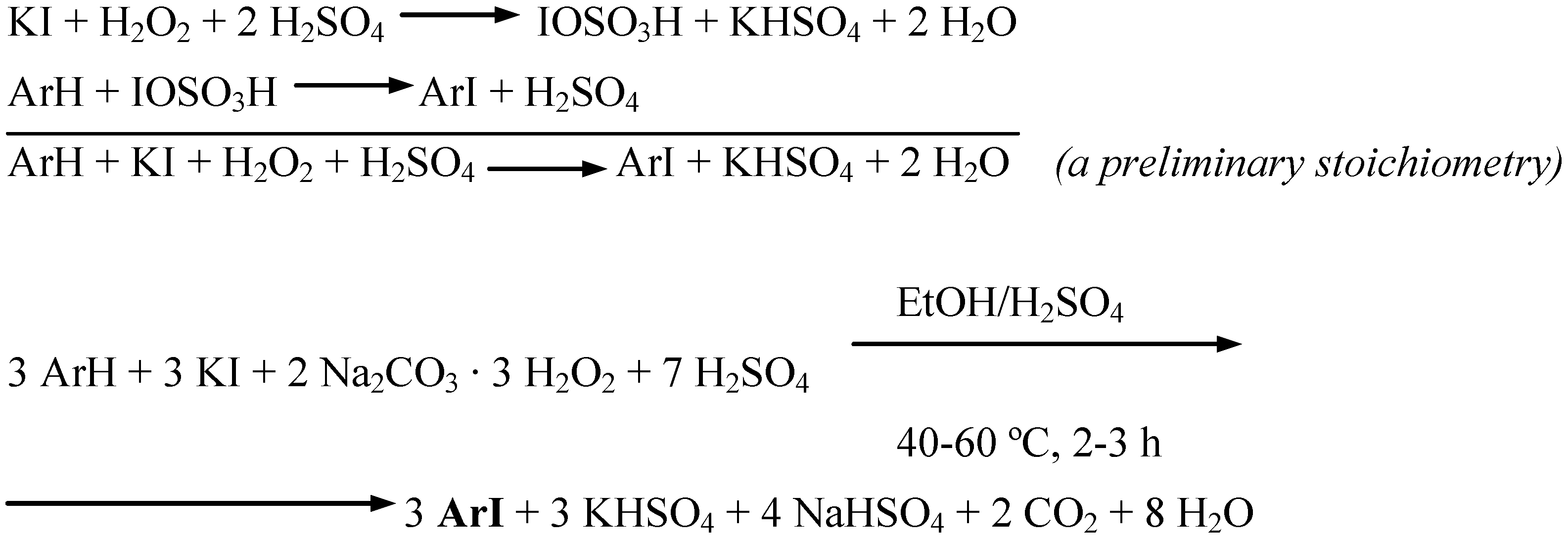{kind=link}
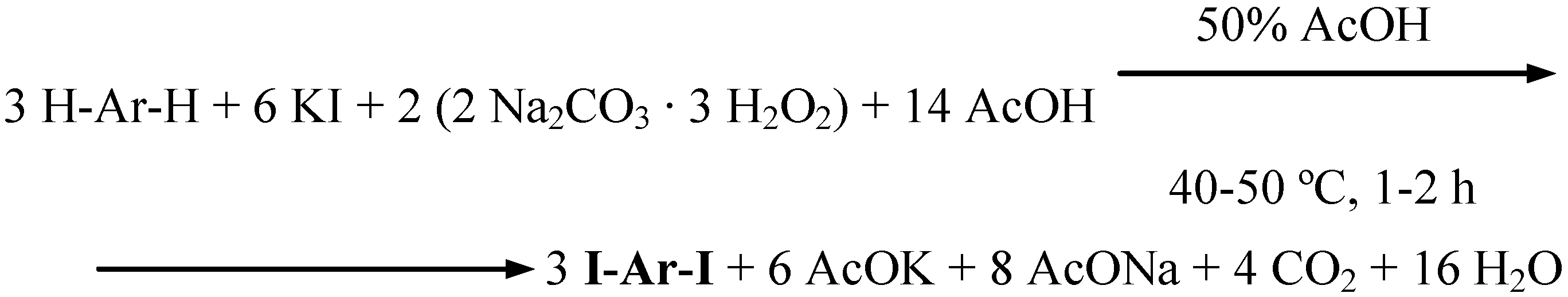{kind=link}
| Substrate | Procedure | Product | Yield (%)a | Mp (°C) (S),b or bp (°C/mmHg); |
|---|---|---|---|---|
| Lit. [12] mp (°C), or bp (°C/mmHg) | ||||
| PhNH2 | 1 | 4-IC6H4NH2 | 68 | 63−65 (H); 63−65 |
| PhNH2 | 1 | 2,4-I2C6H3NH2 | 85 | 93−94 (H); 95−96 |
| 4-IC6H4NH2 | 1 | 2,4-I2C6H3NH2 | 78 | 96−97 (H); 95−96 |
| 2-BrC6H4NH2 | 1 | 2-Br-4-IC6H3NH2 | 67 | 71−74 (Hp); 71−72 |
| 2-MeC6H4NH2 | 1 | 4-I-2-MeC6H3NH2 | 86 | 86−87 (H); 88 |
| PhNMe2 | 2 | 4-IC6H4NMe2 | 60 | 81−83 (E); 82 |
| 2-ClC6H4NH2 | 2 | 2-Cl-4-IC6H3NH2 | 73 | 60−61 (H); 62−63 |
| 2-BrC6H4NH2 | 2 | 2-Br-4-IC6H3NH2 | 62 | 70−73 (Hp); 71−72 |
| 2-MeC6H4NH2 | 2 | 4-I-2-MeC6H3NH2 | 48 | 87−88 (H); 86−88 |
| PhH | 3 | PhI | 40 | bp 76−78/20; bp 78-80/25 [5] |
| PhH | 3 | 1,4-I2C6H4 | 83 | 128−130 (L); 129 |
| 4-O2NC6H4Me | 3 | 2-I-4-O2NC6H3Me | 75 | 51−52 (N); 53−54 |
| 4-O2NC6H4OMe | 3 | 2-I-4-O2NC6H3OMe | 92 | 95−96 (L); 97 |
| 4-MeOC6H4CO2Me | 3 | 3-I-4-MeOC6H4CO2Me | 85 | 93−95 (N); 95−97 |
| PhNHCOMe | 3 | 4-IC6H4NHCOMe | 62 | 183−185 (E); 184 |
| PhCl | 3 | 4-ClC6H4I | 80 | 55−56 (E); 57 |
| PhBr | 3 | 4-BrC6H4I | 68 | 91−92 (L); 91−92 |
| uracil | 3 | 5-iodouracil | 84 | 276−276.5 (E); 276−278 |
| 4-RC6H4CO2Hc | 3 | 3-I-4-RC6H3CO2Hc | 92 | 238−239 (W); 230 |
| PhH | 4 | 1,4-I2C6H4 | 83 | 128−130 (L); 129 |
| PhI | 4 | 1,4-I2C6H4 | 94 | 128−129 (L); 129 |
| PhCO2H | 4 | 3-IC6H4CO2H | 93 | 185−187 (C); 187−188 |
| 4-MeC6H4CO2H | 4 | 3-I-4-MeC6H3CO2H | 79 | 208−209 (C); 210−212 |
| PhCO2Me | 4 | 3-IC6H4CO2Me | 60 | 52−53 (L); 54−55 |
| 4-O2NC6H4Me | 4 | 2-I-4-O2NC6H3Me | 87 | 51−53 (N); 53−54 |
| PhCOPh | 4 | 3-IC6H4COC6H4I-3’ | 51 | 140−142 (A); 141−143 [5] |
| C6H5OMe | 5 | 4-IC6H4OMe | 64 | 50−51 (H); 51−52 |
| 1-MeOC10H7 | 5 | 4-I-1-MeOC10H6 | 68 | 50−51 (E); 52−53 [11] |
| 2-MeOC10H7 | 5 | 1-I-2-MeOC10H6 | 62 | 82−84 (E); 82−84 [11] |
| PhNH2 | 5 | 4-IC6H4NH2 | 53 | 60−62 (H); 63−65 |
| 2-O2NC6H4OH | 6 | 2,4-I2-6-O2NC6H2OH | 80 | 93−94 (E); 94 [13] |
| 4-O2NC6H4OH | 6 | 2,6-I2-4-O2NC6H2OH | 72 | 149−150 (E); 150−151 [11] |
| 4-CH3C6H4OH | 6 | 2,6-I2-4-CH3C6H2OH | 83 | 58−59 (E); 55−58 [14] |
| 8-hydroxyquinoline | 6 | 5,7-diiodo-8-hydroxyquinoline | 85 | 216−217 dec. (E); ca. 214 dec. |
| PhNH2 | 6 | 4-IC6H4NH2 | 77 | 60−62 (H); 63−65 |
| 4-IC6H4NH2 | 6 | 2,4-I2C6H3NH2 | 59 | 95−96 (H); 95−96 |
- a
- Optimized yield of pure isolated product. Satisfactory microanalyses obtained for the purified products: I% ± 0.4; their purities and homogeneities were checked by TLC and 1H and 13C NMR solution spectra (not shown here).
- b
- S = Solvent used for recrystallization. A: acetone; C: CCl4; E: EtOH; H: hexane; Hp: heptane; L: EtOH−H2O (4:1); N: EtOH−H2O (3:2); W: H2O−EtOH (5:1).
- c
- R = 4-MeCONH.
References and Notes
- Reviews on aromatic iodination: Roedig, A. Houben-Weyl, Methoden der organischen Chemie, 4th ed.; Vol. 5/4, Mueller, E., Ed.; Thieme: Stuttgart, 1960; pp. 517–678. [Google Scholar] Merkushev, E. B. Usp. Khim. 1984, 53, 583–594, Russ. Chem. Rev. 1984, 53, 343-353. Merkushev, E. B. Synthesis 1988, 923–937. Sasson, Y. The Chemistry of Halides, Pseudohalides and Azides, Suppl. D1; Patai, S., Rappoport, Z., Eds.; Wiley−Interscience: Chichester, 1995; pp. 535–620. [Google Scholar] Steel, P. G. Rodd’s Chemistry of Carbon Compounds, 2nd ed.; Part 1; Vol. 3, Sainsbury, M., Ed.; Elsevier: Amsterdam, 1996; pp. 178–224. [Google Scholar]
- The latest reviews on organic hypervalent iodine compounds: Varvoglis, A. The Organic Chemistry of Polycoordinated Iodine; VCH: Weinheim, 1992. [Google Scholar] Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, 1997. [Google Scholar] Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. Hypervalent Iodine Chemistry; Wirth, T. (Ed.) Topics in Current Chemistry; Vol. 224, Springer: Berlin, 2003. Stang, P. J. J. Org. Chem. 2003, 68, 2997–3008. Moriarty, R. M. J. Org. Chem. 2005, 70, 2893–2903.
- Skulski, L. Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland (1990-2000). Molecules 2000, 5, 1331–1371. http://www.mdpi.org/molecules/papers/51201331.pdf. [Google Scholar]
- Skulski, L. Novel Easy Preparations of Some Aromatic Iodine(I, III, and V) Reagents, Widely Applied in Modern Organic Synthesis. Molecules 2003, 8, 45–52. http://www.mdpi.org/molecules/papers/80100045.pdf. [Google Scholar]
- Lulinski, P.; Kryska, A.; Sosnowski, M.; Skulski, L. Eco-friendly Oxidative Iodination of Various Arenes with a Urea – Hydrogen Peroxide Adduct (UHP) as the Oxidant. Synthesis 2004, 441–445, We presented there three eco-friendly procedures for the oxidative iodination of both activated and deactivated arenes, using UHP as the oxidant. [Google Scholar]
- Reviews on the uses of a urea-hydrogen peroxide adduct (UHP) in organic synthesis: Heaney, H. Aldrichim. Acta 1993, 26, 35–45. Heaney, H. Organic Peroxygen Chemistry; Herrmann, W. A., Ed.; Topics in Current Chemistry; Vol. 164, Springer: Berlin, 1993; pp. 1–19. [Google Scholar]
- Reviews on the uses of sodium perborate (SPB) and sodium percarbonate (SPC) in organic synthesis: Muzart, J. Synthesis 1995, 1325–1347. McKillop, A.; Sanderson, W. R. Tetrahedron 1995, 51, 6145–6166. McKillop, A.; Sanderson, W. R. J. Chem. Soc., Perkin Trans. 1 2000, 471–476. SPB in the presence of a sodium tungstate catalyst was shown to be a cheap oxidant for the iodination of aromatic amides using KI as the source of iodine(I) species; see: Beinker, P.; Hanson, J. R.; Meindl, N.; Rodriguez Medina, I. C. J. Chem. Res. (S) 1998, 204–205.
- Aldrich Advancing Science; Aldrich: Milwaukee, 2005–2006; The following prices are given therein: (a) Sodium percarbonate (25% H2O2), 13.10 €/500 g. (b) Urea-hydrogen peroxide addition compound (98%), 161.60 €/500 g. (c) Sodium perborate monohydrate, 83.60 €/500 g. (d) Sodium perborate tetrahydrate, 26.80 €/500 g.
- The varied amounts of conc. H2SO4 added to the reaction mixtures (established experimentally and given in the experimental section for each of the substrates iodinated) clearly depended on the relative reactivities of the arenes investigated. The more deactivated the arene, the more conc. H2SO4 had to be added to catalyze better the oxidative iodination reaction. For more details see Ref. 3, pp. 1334−1337.
- Such strongly hygroscopic compounds as I2(SO4)3, I(OSO3H)3, ArISO4, ArI(OSO3H)2 are stable under strongly acidic and anhydrous conditions. For more details see the following review: Kasumov, T. M.; Koz’min, A. S.; Zefirov, N. S. The Chemistry of Inorganic Sulfates and Sulfonates of Polyvalent Iodine. Usp. Khim. 1997, 66, 936–952, Russ. Chem. Rev. 1997, 66, 843-857. [Google Scholar]
- Iskra, J.; Stavber, S.; Zupan, M. Nonmetal-catalyzed Iodination of Arenes with Iodide and Hydrogen Peroxide. Synthesis 2004, 1869–1873. [Google Scholar] [CrossRef] [Green Version]
- Dictionary of Organic Compounds, 6th ed.; Chapman & Hall: London, 1996.
- Hodgson, H. H.; Smith, E. W. J. Chem. Soc. 1932, 503–505.
- Bell, N. V.; Bowman, W. R.; Coe, P. F.; Turner, A. T.; Whybrow, D. Can. J. Chem. 1997, 75, 873–889.
- Sample Availability: Contact the authors.
© 2005 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Zielinska, A.; Skulski, L. Eco-friendly Oxidative Iodination of Various Arenes with Sodium Percarbonate as the Oxidant†. Molecules 2005, 10, 1307-1317. https://doi.org/10.3390/10101307
AMA Style
Zielinska A, Skulski L. Eco-friendly Oxidative Iodination of Various Arenes with Sodium Percarbonate as the Oxidant†. Molecules. 2005; 10(10):1307-1317. https://doi.org/10.3390/10101307
Chicago/Turabian StyleZielinska, Agnieszka, and Lech Skulski. 2005. "Eco-friendly Oxidative Iodination of Various Arenes with Sodium Percarbonate as the Oxidant†" Molecules 10, no. 10: 1307-1317. https://doi.org/10.3390/10101307