Valence Topological Charge-Transfer Indices for Reflecting Polarity: Correction for Heteromolecules

Institut Universitari de Ciència Molecular, Universitat de València, Spain
Molecules 2005, 10(2), 334-345; https://doi.org/10.3390/10020334
Submission received: 29 March 2004
/
Accepted: 10 May 2004
/
Published: 28 February 2005
(This article belongs to the Special Issue Sulfur-Nitrogen Heterocycles)
Abstract
:Valence topological charge-transfer (CT) indices are applied to the calculation of dipole moments μ. The μ calculated by algebraic and vector semisums of the CT indices are defined. The model is generalized for molecules with heteroatoms and corrected for sp3-heteromolecules. The ability of the indices for the description of the molecular charge distribution is established by comparing them with μ of the valence-isoelectronic series of cyclopentadiene, benzene and styrene. Two CT indices, μvec (vector semisum of vertex-pair μ) and μvecV (valence μvec) are proposed. The μvecV behaviour is intermediate between μvec and μexperiment. The correction is produced in the correct direction. The best results are obtained for the greatest group. Inclusion of the heteroatom in the π-electron system is beneficial for the description of μ, owing to either the role of additional p and/or d orbitals provided by the heteroatom or the role of steric factors in the π-electron conjugation. The steric effect is almost constant along the series and the dominating effect is electronic. Inclusion of the heteroatom enhances μ, which can improve the solubility of the molecule. For heteroatoms in the same group, the ring size and the degree of ring flattering are inversely proportional to their electronegativity.
Introduction
Homo and heterocycles were studied as models of fluorescers, organic conducting polymers or nonlinear optical (NLO) materials. New fluorescers contain heteroaromatic components. Some heterocycles recur often in industrial fluorescers. They do not fluoresce themselves, but have a fluorescence enhancing effect when coupled to conjugated systems. Reiser et al. measured the absorption and emission spectra, and fluorescence yields of aromatic benzoxazole derivatives [1]. Lippert et al. reviewed the photophysics of internal twisting [2]. Dey and Dogra measured and calculated the solvatochromism and prototropism in 2-(aminophenyl)benzothiazoles [3]. Catalán et al. studied the role of the torsion of the phenyl moiety in the mechanism of stimulated ultraviolet light generation in 2-phenylbenzazoles [4]. Levitus et al. performed photophysical measurements and semiempirical calculations with 1,4-bis(phenylethynyl)benzene [5]. Organic conducting polymers have a highly anisotropic quasi-one-dimensional (quasi-1D) structure similar to that of charge-transfer (CT) salts [6]. In the conducting state, both materials are ionic. In CT complexes conductivity is greater along the stacking direction, while in conducting polymers conductivity is higher along the chain direction. In these polymers, the chainlike structure leads to strong coupling of the electronic states to conformational excitations peculiar to 1D systems. The relatively weak interchain binding allows diffusion of dopant molecules into the structure, while the strong intrachain C–C bonds maintain the integrity of the polymer. The modulation of the electronic properties of conjugated polymers was studied through design of polymer backbone [7].
The search for NLO organic materials with large values of the second hyperpolarizability (γ) attracted interest from the experimental and theoretical points of view [8,9]. Morley et al. calculated the first hyperpolaizability (β) of S-heteromolecules [10]. Zhao et al. computed γ for modified benzothiazoles, benzoxazoles and benzimidazoles [11]. Meyers et al. calculated the geometries and electronic and NLO properties of CT molecules based on the 2-methylene-2H-pyrrole repeating unit [12]. Li et al. computed structure–performance characteristics for β and γ of π-conjugated organic chromophores with the Pariser–Parr–Pople model [13]. Yeates et al. analyzed (X-ray) and calculated 2-(2-benzothiazolyl)-1-(2-thienyl)ethene as a model for high γ [14]. Gao et al. studied the effect of conjugated length on the computed β of organic molecules [15]. Tomonari et al. calculated the simplified sum-over-states and missing-orbital analysis on β and γ of benzene derivatives [16]. Glaser and Chen computed asymmetrization effects on structures of dipolar donor–acceptor-substituted molecular organic NLO materials [17]. Raptis et al. calculated the polarizability (α), β and γ of polysulfanes [18]. Rao and Bhanuprakash computed donor and acceptor organic molecules separated by a saturated C–C σ bond, which show large β [19]. Nakano et al. calculated γ of tetrathiapentalene and tetrathiafulvalene [20]. Cheng et al. computed α and β of H-silsesquioxanes [21]. Levitus et al. analyzed and calculated the photophysics of 1,4-bis(phenylethynyl)benzene [22] and 1,4-diethynyl- 2-fluorobenzene [23] to demarcate the effects of chromophore aggregation and planarization in poly(phenyleneethynylene)s. Öberg et al. computed β and γ of organic compounds [24].
Organic electronic materials are conjugated solids where both optical absorption and charge transport are dominated by partly delocalized π and π* orbitals [25]. Organic photovoltaic materials differ from inorganic semiconductors in the following important respects. (1) Photogenerated excitations (excitons) are strongly bound and do not spontaneously dissociate into charge pairs. This means that carrier generation does not necessarily result for the absorption of light. (2) Charge transport proceeds by hopping between localized states, rather than transport within a band, and mobilities are low. (3) The spectral range of optical absorption is relatively narrow compared to the solar spectrum. (4) Absorption coefficients are high so that high optical densities can be achieved, at peak wavelength, with films less than 100nm thick. (5) Many materials are susceptible to degradation in the presence of oxygen or water. (6) As 1D semiconductors, their electronic and optical properties can be highly anisotropic. These properties impose some constraints on organic photovoltaic devices. (1) A strong driving force such as an electric field should be present to break up the photogenerated excitons. (2) Low charge carrier mobilities limit the useful thickness of devices. (3) Limited light absorption across the solar spectrum limits the photocurrent. (4) Very thin devices mean interference effects can be important. (5) Photocurrent is sensitive to temperature through hopping transport.
In earlier publications, α of benzothiazole (A)–benzobisthiazole (B) A–Bn–A (n≤13) oligomers was calculated and extrapolated to n→∞ [26]. Torsional effects were analyzed [27,28]. CT indices were brought to the calculation of the dipole moment μ of hydrocarbons [29]. The model was extended to heteroatoms [30]. An index inspired by plastic evolution improved the results [31,32]. The method was applied to the valence-isoelectronic series of benzene and styrene (2–4 molecules) [33,34]. This study presents a reparametrization of the method for sp3-heteromolecules. The next section introduces CT indices. Following that, the correction for sp3-heteromolecules is presented. Next, the results are discussed. The last section summarizes the conclusions.
Topological Charge-Transfer Indices
The most important matrices that delineate the labelled chemical graph are the adjacency (A) [35] and the distance (D) matrices, wherein Dij=lij if i=j, “0” otherwise; lij is the shortest edge count between vertices i and j [36]. In A, Aij=1 if vertices i and j are adjacent, “0” otherwise. The D[-2] matrix is the matrix whose elements are the squares of the reciprocal distances Dij-2. The intermediate matrix M is defined as the matrix product of A by D[-2]:
The charge-transfer matrix C is defined as C = M – MT, where MT is the transpose of M [37]. By agreement, Cii=Mii. For i≠j, the Cij terms represent a measure of the intramolecular net charge transferred from atom j to i. The topological CT indices Gk are described as the sum, in absolute value, of the Cij terms defined for the vertices i,j placed at a topological distance Dij equal to k
where N is the number of vertices in the graph, Dij are the entries of the D matrix, and δ is the Kronecker δ function, being δ=1 for i=j and δ=0 for i≠j. Gk represents the sum of all the Cij terms, for every pair of vertices i and j at topological distance k. Other topological CT index, Jk, I defined as:
M = AD[-2]
This index represents the mean value of the CT for each edge, since the number of edges for acyclic compounds is N–1.
The algebraic semisum CT index μalg is defined as
where Ce is the Cij index for vertices i and j connected by edge e [29]. The sum extends for all pairs of adjacent vertices in the molecular graph and μalg is a graph invariant. An edge-to-edge analysis of μ suggests that each edge dipole moment μe connecting vertices i and j can be evaluated from the corresponding edge Ce index as
Each edge dipole can be associated with a vector μe in space. This vector has magnitude |μe|, lies in the edge e connecting vertices i and j, and its direction is from j to i. The molecular dipole moment vector μ results the vector sum of the edge dipole moments as
summed for all the m edges in the molecular graph. The vector semisum CT indexμvec is defined as the module of μ:
and μvec is a graph invariant.
When heteroatoms are present, some way of discriminating atoms of different kinds needs to be considered [38]. In valence CT indices terms, the presence of each heteroatom is taken into account by introducing its electronegativity value in the corresponding entry of the main diagonal of the adjacency matrix A. For each heteroatom X, its entry Aii is redefined as
to give the valence adjacency AV matrix where χX and χC are the electronegativities of heteroatom X and carbon, respectively, in Pauling units. Notice that the subtractive term keeps AiiV=0 for the C atom (Equation 5). Moreover, the multiplicative factor reproduces AiiV=2.2 for O, which was taken as standard. From the valence AV, MV and CV matrices, μalgV, μvecV and topological CT indices GkV and JkV can be calculated by following the former procedure with the AV matrix. The CiiV, GkV, JkV, μalgV and μvecV descriptors are graph invariants. The main difference between μvec and μvecV is that μvec is sensitive only to the steric effect of the heteroatoms, while μvecV is sensitive to both electronic and steric effects.
Correction for sp3-Heteroatom-Containing Compounds
Kubinyi showed that the poor hydrogen-bond-formation capacity of the sp3-oxygen atoms that are directly linked to an sp2-carbon atom (like in esters, aromatic ethers and furans) is also reflected by a significant decrease of their polarity (MedChem database 1-octanol–water partition coefficient, P) in going from aliphatic to araliphatic and to aromatic ethers R–O–R’ (Table 1) [39]. Therefore, in this study it is suggested to halve the factor in Equation (5) as
for sp3-X (–X–), X = O. Table 1 gives the molecular dipole moments μ for hydrocarbons and ethers calculated with different charge-transfer indices. The polarity decrease is also reflected by a significant decrease of the differential dipole moment (μether – μhydrocarbon) denoted as Δ(O – CH2). The Δ(O – CH2) μexperiment decreases with minus Δ(O – CH2) logP. The Δ(O – CH2) μvec does not show this diminution, while Δ(O – CH2) μvecV gives very great values. However, Δ(O – CH2) μvecV,corrected is of the same order of magnitude as both μexperiment and μMOPAC-AM1 references. As similar effects were shown for sp3-Si, P, Ge, As, Sn, Sb, Pb and Bi heteromolecules [34], Equation (6) is used for all sp3-X (–X–), X = O, Si, P, S, Ge, As, Se, Sn, Sb, Te, Pb, Bi, Po.

{kind=link}
{kind=link}
{kind=link}
| Method | Compound | X = –CH2– | X = –O– | Δ(O – CH2) | Δ(O–CH2) log Pa |
|---|---|---|---|---|---|
| Vector semisum | Et–X–Et | 0.407 | 0.436 | 0.029 | – |
| Phe–X–Et | 0.739 | 0.659 | -0.080 | – | |
| Phe–X–Phe | 0.427 | 0.333 | -0.094 | – | |
| Valence vector semisum | Et–X–Et | 0.407 | 2.854 | 2.447 | – |
| Phe–X–Et | 0.739 | 2.621 | 1.882 | – | |
| Phe–X–Phe | 0.427 | 2.742 | 2.315 | – | |
| Corrected valence vectorsemisum | Et–X–Et | 0.407 | 1.209 | 0.802 | – |
| Phe–X–Et | 0.739 | 1.211 | 0.472 | – | |
| Phe–X–Phe | 0.427 | 1.204 | 0.777 | – | |
| Experimentb | Et–X–Et | 0.087c | 1.170 | 1.083 | -2.50 |
| Phe–X–Et | 0.350 | 1.410 | 1.060 | -1.21 | |
| Phe–X–Phe | 0.260 | 1.150 | 0.890 | 0.07 | |
| AM1 | Et–X–Et | 0.006 | 1.246 | 1.240 | – |
| Phe–X–Et | 0.257 | 1.264 | 1.007 | – | |
| Phe–X–Phe | 0.080 | 1.252 | 1.172 | – |
a P is the 1-octanol–water partition coefficient.
b Reference 40.
c Gaussian-2 composite ab initio method calculation taken from Reference 41.
Calculation Results and Discussion
The molecular CT indices Gk, Jk, GkV and JkV (with k<6) are listed in Table 2 for the valence-isoelectronic series of benzene (C6H6). As one might have expected, all the molecules show the same set of Gk (and, consequently, Jk) values. For instance, G1 is related to the degree of branching and G2 is related to the number of unsaturations in the molecule, which are constant throughout the series. On the other hand, GkV, which also depends on the electronegativity of the heteroatom through Equations (5–6), is influenced, in general, by the substitution.
In particular, G1V is related to the absolute differential electronegativity of the heteroatom |χX–χC| in the molecule. However, an exception occurs: G2V (and, as a result, J2V) is equal throughout the series. C5H6Si, C5H6Ge, C5H6Sn and C5H6Pb show the same results for all GkV and JkV. This is due to the fact that Si, Ge, Sn and Pb have the same electonegativity (χSi=χGe=χSn=χPb=1.8). The same happens for C5H5Sb and C5H5Bi (χSb=χBi=1.9). Although in pyridine and C5H5As, the N and As atoms have the same absolute differential electronegativity |χN–χC|=|χAs–χC|=0.5, pyridine is calculated by Equation (5) while C5H5As, by Equation (6), and so G2V(pyridine) = 2G2V(C5H5As).
Table 2.
Charge indices up to the fifth order for the valence-isoelectronic series of benzene.a
| Molecules | N | G1 | G2 | J1 | J2 |
|---|---|---|---|---|---|
| all molecules | 6 | 0.0000 | 5.3333 | 0.0000 | 1.0667 |
| Molecules | G1V | G3V | J1V | J3V |
|---|---|---|---|---|
| benzene | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| pyridine | 2.2000 | 0.1222 | 0.4400 | 0.0244 |
| C5SiH6 | 1.5400 | 0.0856 | 0.3080 | 0.0171 |
| C5PH5 | 0.8800 | 0.0489 | 0.1760 | 0.0098 |
| C5GeH6 | 1.5400 | 0.0856 | 0.3080 | 0.0171 |
| C5AsH5 | 1.1000 | 0.0611 | 0.2200 | 0.0122 |
| C5SnH6 | 1.5400 | 0.0856 | 0.3080 | 0.0171 |
| C5SbH5 | 1.3200 | 0.0733 | 0.2640 | 0.0147 |
| C5PbH6 | 1.5400 | 0.0856 | 0.3080 | 0.0171 |
| C5BiH5 | 1.3200 | 0.0733 | 0.2640 | 0.0147 |
a Gi, Ji (i > 2), GiV, JiV (i > 3) are zero for all the entries; G2V = 5.3333, J2V = 1.0667.
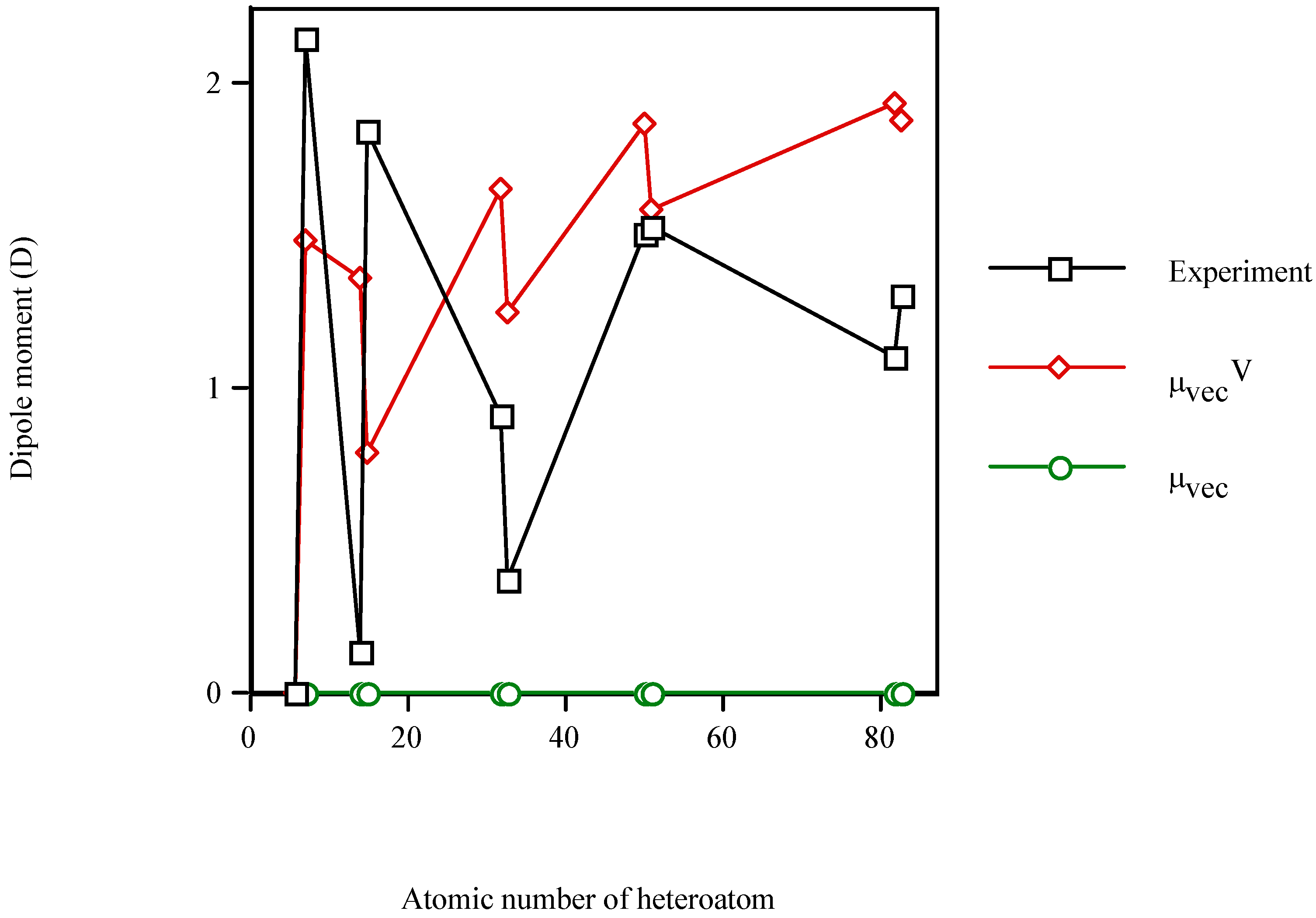
Figure 1 illustrates the increment in the dipole moment of benzene in the presence of the heteroatom. The calculated μvecV is of the same order of magnitude as μexperiment, while the calculated μvec=0 remains constant. Since μvec is sensitive to the steric effect but not to the electronic effect of the heteroatom, it is clear that the electronic effect (μvecV) dominates over the steric one (μvec). In particular, the best results are obtained for the fourth long-period (Sn–Sb) and for the group-V heteromolecules.
Figure 1.
Dipole moment of the valence-isolectronic series of benzene vs. the atomic number of the heteroatom. Experimental data from Reference 40. Points with Z = 14, 15, 32, 33, 50, 51, 82 and 83 are AM1 calculations.
Figure 1.
Dipole moment of the valence-isolectronic series of benzene vs. the atomic number of the heteroatom. Experimental data from Reference 40. Points with Z = 14, 15, 32, 33, 50, 51, 82 and 83 are AM1 calculations.

Table 3.
Charge indices up to the fifth order for the valence-isoelectronic series of styrene.
| Molecule | N | G1 | G2 | G3 | G4 | G5 |
|---|---|---|---|---|---|---|
| all molecules | 8 | 1.0000 | 6.8889 | 0.4375 | 0.2133 | 0.0625 |
| Molecules | J1 | J2 | J3 | J4 | J5 |
|---|---|---|---|---|---|
| all molecules | 0.1429 | 0.9841 | 0.0625 | 0.0305 | 0.0089 |
| Molecules | G1V | G2V | G3V | G4V | G5V |
|---|---|---|---|---|---|
| styrene | 1.0000 | 6.8889 | 0.4375 | 0.2133 | 0.0625 |
| benzaldimine | 1.6000 | 7.1639 | 0.5569 | 0.2708 | 0.0185 |
| benzaldehyde | 2.7000 | 7.4389 | 0.8014 | 0.4083 | 0.0255 |
| C6H5–CH=SiH2 | 2.5400 | 6.5039 | 0.5297 | 0.3436 | 0.1241 |
| C6H5–CH=PH | 1.8800 | 6.6689 | 0.4375 | 0.2611 | 0.0977 |
| thiobenzaldehyde | 1.0000 | 6.8889 | 0.4375 | 0.2133 | 0.0625 |
| C6H5–CH=GeH2 | 2.5400 | 6.5039 | 0.5297 | 0.3436 | 0.1241 |
| C6H5–CH=AsH | 2.1000 | 6.6139 | 0.4375 | 0.2886 | 0.1065 |
| C6H5–CH=Se | 1.2200 | 6.8339 | 0.4375 | 0.2133 | 0.0713 |
| C6H5–CH=SnH2 | 2.5400 | 6.5039 | 0.5297 | 0.3436 | 0.1241 |
| C6H5–CH=SbH | 2.3200 | 6.5589 | 0.4808 | 0.3161 | 0.1153 |
| C6H5–CH=Te | 1.8800 | 6.6689 | 0.4375 | 0.2611 | 0.0977 |
| C6H5–CH=PbH2 | 2.5400 | 6.5039 | 0.5297 | 0.3436 | 0.1241 |
| C6H5–CH=BiH | 2.3200 | 6.5589 | 0.4808 | 0.3161 | 0.1153 |
| C6H5–CH=Po | 2.1000 | 6.6139 | 0.4375 | 0.2886 | 0.1065 |
| Molecules | J1V | J2V | J3V | J4V | J5V |
|---|---|---|---|---|---|
| styrene | 0.1429 | 0.9841 | 0.0625 | 0.0305 | 0.0089 |
| benzaldimine | 0.2286 | 1.0234 | 0.0796 | 0.0387 | 0.0026 |
| benzaldehyde | 0.3857 | 1.0627 | 0.1145 | 0.0583 | 0.0036 |
| C6H5–CH=SiH2 | 0.3629 | 0.9291 | 0.0757 | 0.0491 | 0.0177 |
| C6H5–CH=PH | 0.2686 | 0.9527 | 0.0625 | 0.0373 | 0.0140 |
| thiobenzaldehyde | 0.1429 | 0.9841 | 0.0625 | 0.0305 | 0.0089 |
| C6H5–CH=GeH2 | 0.3629 | 0.9291 | 0.0757 | 0.0491 | 0.0177 |
| C6H5–CH=AsH | 0.3000 | 0.9448 | 0.0625 | 0.0412 | 0.0152 |
| C6H5–CH=Se | 0.1743 | 0.9763 | 0.0625 | 0.0305 | 0.0102 |
| C6H5–CH=SnH2 | 0.3629 | 0.9291 | 0.0757 | 0.0491 | 0.0177 |
| C6H5–CH=SbH | 0.3314 | 0.9370 | 0.0687 | 0.0452 | 0.0165 |
| C6H5–CH=Te | 0.2686 | 0.9527 | 0.0625 | 0.0373 | 0.0140 |
| C6H5–CH=PbH2 | 0.3629 | 0.9291 | 0.0757 | 0.0491 | 0.0177 |
| C6H5–CH=BiH | 0.3314 | 0.9370 | 0.0687 | 0.0452 | 0.0165 |
| C6H5–CH=Po | 0.3000 | 0.9448 | 0.0625 | 0.0412 | 0.0152 |
The molecular CT indices Gk, Jk, GkV and GkV for the valence-isoelectronic series of styrene (C6H5–CH=CH2) are collected in Table 3. As expected, all the molecules show the same set of Gk and Jk values. However, GkV are influenced by the atomic number of the heteroatom. In particular, the results for thiobenzaldehyde are equal to those for styrene. This is because the electronegativity for the S atom has been taken equal to that of C (χS=χC=2.5). The same happens for the Si/Ge/Sn/Pb (χSi=χGe=χSn=χPb=1.8), P/Te (χP=χTe=2.1), As/Po (χAs=χPo=2.0) and Sb/Bi compounds (χSb=χBi=1.9).
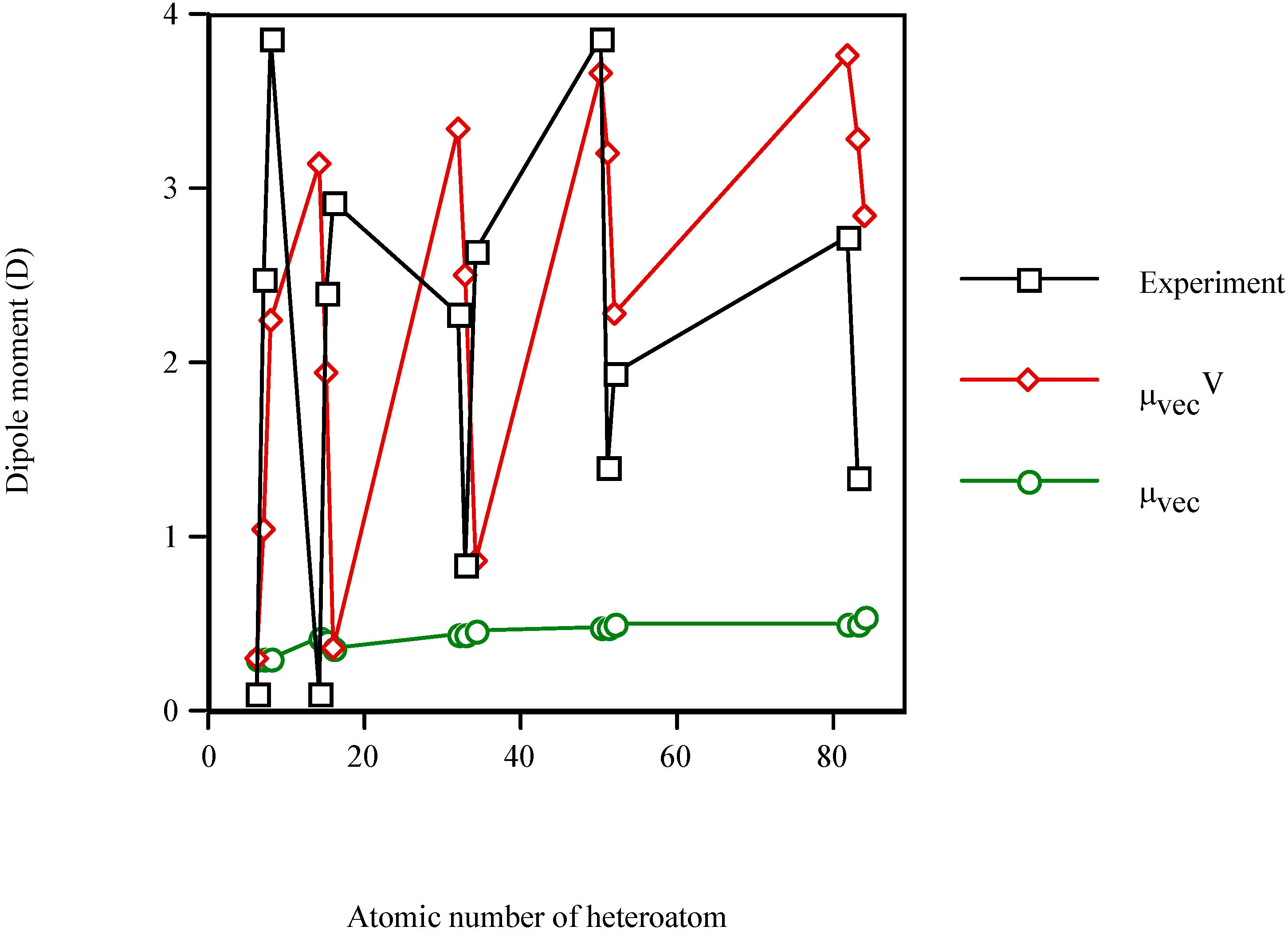
Figure 2 shows the increase in the dipole moment of the valence-isoelectronic series of styrene when the heteroatom is present. Again, μexperiment and μvecV vary in a similar fashion, while μvec remains almost constant (μvec~0.43D for the three groups IV–VI). The electronic effect of the heteroatom (μvecV) dominates, in general, over the steric one (μvec). In particular, for thiobenzaldehyde (Z=16) the result of μvecV = μvec (because χS=χC) should be taken with care. It is an artefact of the model for S-heteromolecules. Furthermore, the best results are obtained, in general, for the fourth long-period (Sn–Te) and for the group-VI heteromolecules.
Figure 2.
Dipole moment of the valence-isolectronic series of styrene vs. the atomic number of the heteroatom. Point with Z = 6 from Reference 42; Z = 7, 14–16, 32–34, 51 and 52 are computed with AM1; Z = 8 from Reference 43; Z = 50, 82 and 83 are PM3 calculations.
Figure 2.
Dipole moment of the valence-isolectronic series of styrene vs. the atomic number of the heteroatom. Point with Z = 6 from Reference 42; Z = 7, 14–16, 32–34, 51 and 52 are computed with AM1; Z = 8 from Reference 43; Z = 50, 82 and 83 are PM3 calculations.

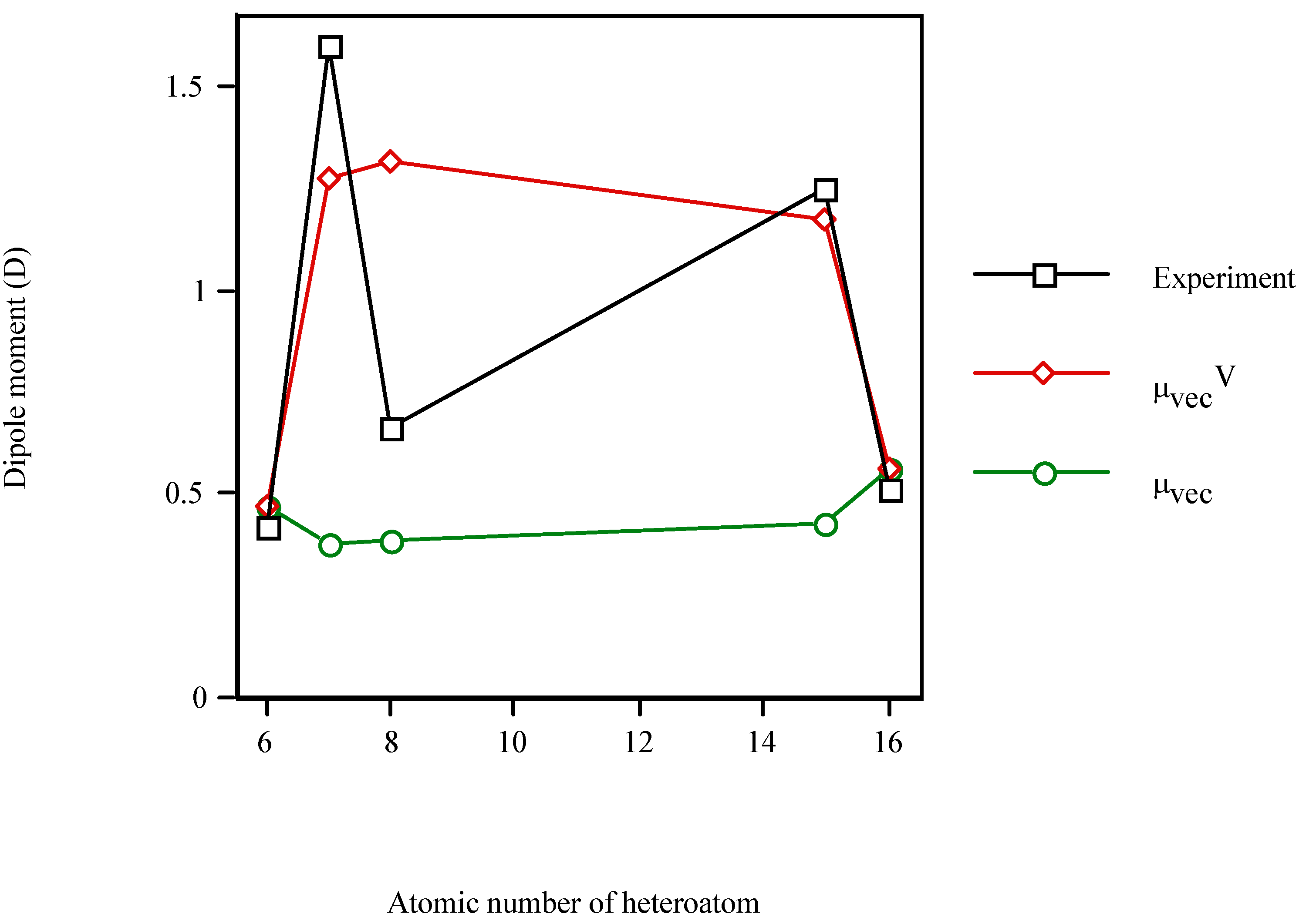
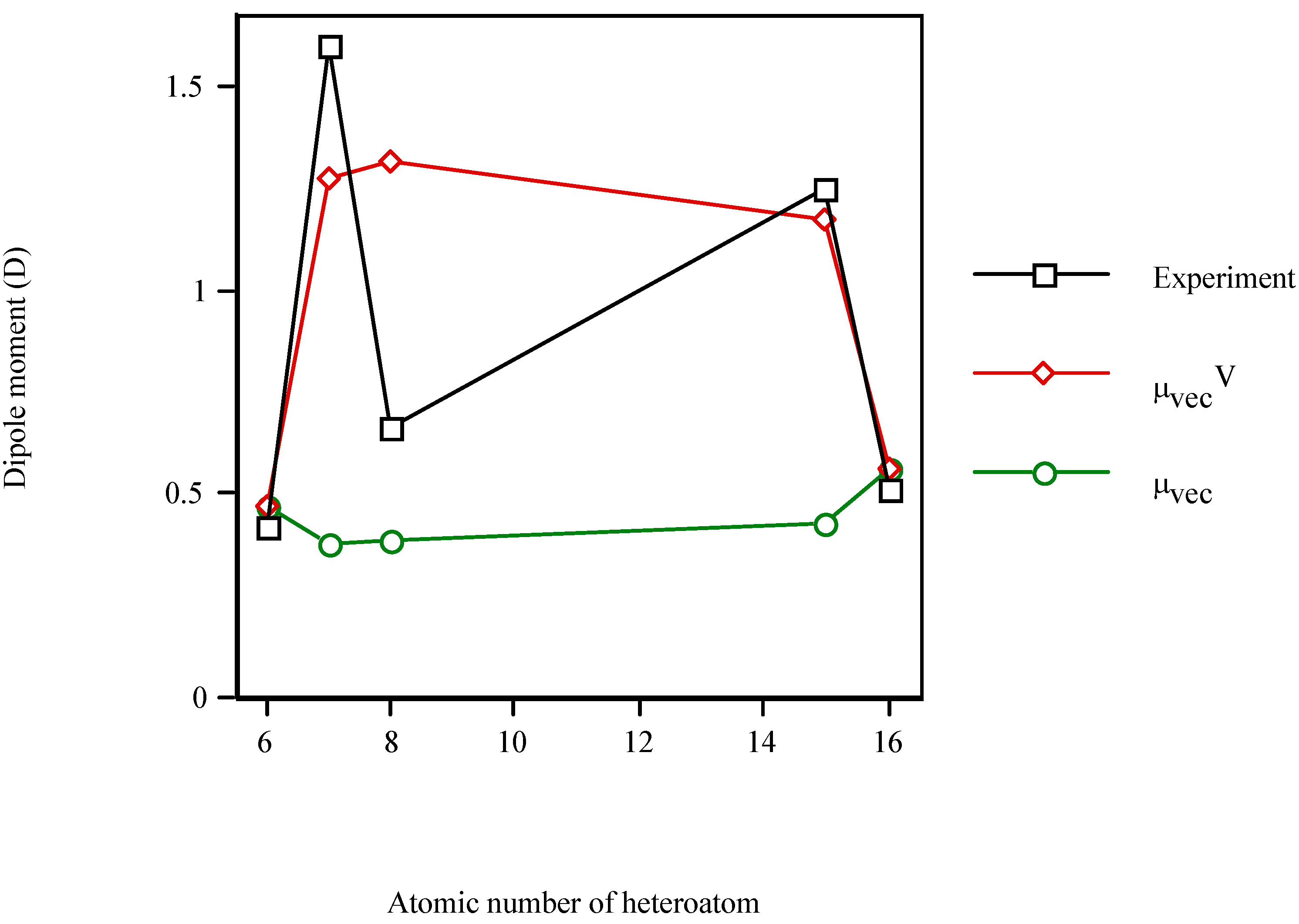
In order to test the model for other S-containing heterocycle, Figure 3 displays the increase in the dipole moment of the valence-isoelectronic series of cyclopentadiene when the heteroatom is present. Once more, μexperiment and μvecV vary in a similar fashion while μvec remains almost constant (μvec~0.45D). The electronic effect of the heteroatom (μvecV) dominates over the steric one (μvec). In particular, for thiophene (Z=16) μvecV = μvec (because χS=χC). However, the μvecV relative error for thiophene (10%) is even smaller than for cyclopentadiene (12%).
Figure 3.
Dipole moment of the valence-isolectronic series of cyclopentadiene vs. the atomic number of the heteroatom. Point with Z = 15 is AM1 calculation.
Figure 3.
Dipole moment of the valence-isolectronic series of cyclopentadiene vs. the atomic number of the heteroatom. Point with Z = 15 is AM1 calculation.
Conclusions
The following conclusions can be made from this study:
- The behaviour of μvecV is intermediate between μvec and μexperiment and so the correction introduced with respect to μvec is produced in the correct direction. The best results are obtained for the greatest group that can be studied.
- Inclusion of the heteroatom in the π-electron system is beneficial for the description of the dipole moment, owing to either the role of additional p and/or d orbitals provided by the heteroatom or the role of steric factors in the π-electron conjugation. The analysis of both electronic and steric factors in μ caused by the presence of the heteroatom shows that the electronic factor dominates over the steric one. Work is in progress on the calculation of the dipole moments of a homologous series of 4-alkylanilines, which are percutaneous enhancers of transdermal-delivery drugs.
- Inclusion of the heteroatom enhances μ with the only exception of the insertion of the Si atom in styrene. In turn, the increase in μ can improve the solubility of the molecule.
- For heteroatoms in the same group of the periodic table, the ring size and the degree of ring flattering are inversely proportional to the electronegativity of the hetetoatom, e.g., cyclopentadienering < C4SiH6 ring < C4GeH6 ring < C4SnH6 ring < C4PbH6 ring and benzenering < C5SiH6 ring < C5GeH6 ring < C5SnH6 ring < C5PbH6 ring.
- Inclusion of the heteroatom increases μ, which is smaller for the benzene than for the styrene series. On going from styrene to C6H5–CH=SnH2, μexperiment increases by a factor of 41. Although there is a minor steric effect (μvec increases by a factor of 1.6), the major effect is electronic (μvecV augments by a factor of 12). From μvec to μvecV the introduced correction is produced in the correct direction. However, the result for thiobenzaldehyde is uncertain. Work is in progress with the correct parameterization of the method for the S atom. On going from cyclopentadiene to pyrrole, μexperiment increases by a factor of 4. Although there is an antagonistic steric effect (in fact μvec decreases), the major effect is electronic (μvecV is trebled).
Acknowledgements
The author acknowledges financial support from the Spanish MCT (Plan Nacional I+D+I, Project No. BQU2001-2935-C02-01) and Generalitat Valenciana (DGEUI INF01-051 and INFRA03-047, and OCYT GRUPOS03-173).
References
- Reiser, A.; Leyshon, L. J.; Saunders, D.; Mijovic, M. V.; Bright, A.; Bogie, J. Fluorescence of Aromatic Benzoxazole Derivatives. J. Am. Chem. Soc. 1972, 94, 2414–2421. [Google Scholar]
- Lippert, E.; Rettig, W.; Bonaçi≈-Kouteck¥, V.; Heisel, F.; Miehé, J. A. Photophysics of Internal Twsting. Adv. Chem. Phys. 1987, 68, 1–173. [Google Scholar]
- Dey, J. K.; Dogra, S. K. Solvatochromism and Prototropism in 2-(Aminophenyl) Benzothiazoles. Bull. Chem. Soc. Jpn. 1991, 64, 3142–3152. [Google Scholar] [CrossRef]
- Catalán, J.; Mena, E.; Fabero, F.; Amat-Guerri, F. The Role of the Torsion of the Phenyl Moiety in the Mechanism of Stimulated Ultraviolet Light Generation in 2-Phenylbenzazoles. J. Chem. Phys. 1992, 96, 2005–2016. [Google Scholar] [CrossRef]
- Levitus, M.; Schmieder, K.; Ricks, H.; Shimizu, K. D.; Bunz, U. H. F.; Garcia-Garibay, M. A. Steps to Demarcate the Effects of Chromophore Aggregation and Planarization in Poly(phenyleneethynylene)s. 1. Rotationally Interrupted Conjugation in the Excited States of 1,4-Bis(phenylethynyl)benzene. J. Am. Chem. Soc 2001, 123, 4259–4265. [Google Scholar]
- Patil, A. O.; Heeger, A. J.; Wudl, F. Optical Properties of Conducting Polymers. Chem. Rev. 1988, 88, 183–200. [Google Scholar] [CrossRef]
- Piqueras, M. C.; Crespo, R.; Tomá, F. Modulation of the Electronic Properties of Conjugated Polymers Through Design of Polymer Backbone. J. Mol. Struct. (Theochem) 1995, 330, 181–185. [Google Scholar]
- Nonlinear Optical Properties of Polymers; Heeger, A. J.; Orenstein, J.; Ulrich, D. R. (Eds.) Materials Research Society Symp. Proc. Vol. 109; MRS: Pittsburgh, 1987.
- Nonlinear Optical and Electroactive Polymers; Prasad, P. N.; Ulrich, D. R. (Eds.) Plenum: New York, 1988.
- Morley, J. O.; Docherty, V. J.; Pugh, D. Non-Linear Optical Properties of Organic Molecules. Part 4. Calculations of the Hyperpolarisability of Sulphur-Containing Systems. J. Chem. Soc., Perkin Trans. 2 1987, 1361–1363. [Google Scholar]
- Zhao, M.; Samoc, M.; Prasad, P. N.; Reinhardt, B. A.; Unroe, M. R.; Prazak, M.; Evers, R. C.; Kane, J. J.; Jariwala, C.; Sinsky, M. Studies of Third-Order Optical Nonlinearities of Model Compounds Containing Benzothiazole, Benzimidazole, and Benzoxazole Units. Chem. Mater. 1990, 2, 670–678. [Google Scholar] [CrossRef]
- Meyers, F.; Adant, C.; Brédas, J. L. Theoretical Investigation of the Electronic and Geometric Structures and Nonlinear Optical Properties of 2H-pyrrole Derivatives. J. Am. Chem. Soc. 1991, 113, 3715–3719. [Google Scholar] [CrossRef]
- Li, D.; Marks, T. J.; Ratner, M. A. Nonlinear Optical Phenomena in Conjugated Organic Chromophores. Theoretical Investigations via a π-Electron Formalism. J. Phys. Chem. 1992, 96, 4325–4336. [Google Scholar]
- Yeates, A. T.; Dudis, D. S.; Resch, T.; Fratini, A. V. Materials Research Society Symp. Proc.; Vol. 247; MRS: Pittsburgh, 1992; pp. 259–264. [Google Scholar]
- Gao, X. L.; Feng, J. K.; Sun, C. C. Effect of Conjugated Length on the Calculated Nonlinear Second-Order Optical Susceptibilities of Some Organic Molecules. Int. J. Quantum Chem. 1992, 42, 1747–1758. [Google Scholar] [CrossRef]
- Tomonari, M.; Ookubo, N.; Takada, T.; Feyereisen, M. W.; Almlöf, J. Simplified Sum-over-States Calculations and Missing-Orbital Analysis on Hyperpolarizabilities of Benzene Derivatives. Chem. Phys. Lett. 1993, 203, 603–610. [Google Scholar] [CrossRef]
- Glaser, R.; Chen, G. S. Asymmetrization Effects on Structures and Populations of the Ground State of Dipolar Donor–Acceptor-Substituted Molecular Organic NLO Materials. J. Comput. Chem. 1998, 19, 1130–1140. [Google Scholar] [CrossRef]
- Raptis, S. G.; Nasiou., S. M.; Demetropoulos, I. N.; Papadopoulos, M. G. Static and Frequency Dependent Polarizabilities and Hyperpolarizabilities of H2Sn. Comput. Chem. 1998, 19, 1698–1715. [Google Scholar] [CrossRef]
- Rao, J. L.; Bhanuprakash, K. Theoretical Studies on the Non-Linear Optical Properties of Some Organic Molecules: Effect of π–σ–π through-Bond Coupling on the First Hyperpolarizability. J. Mol. Struct. (Theochem) 1999, 458, 269–273. [Google Scholar]
- Nakano, M.; Yamada, S.; Yamaguchi, K. On the Second Hyperpolarizabilities γ of Three Charged States of Tetrathiapentalene and Tetrathiafulvalene: A γ Density Analysis. Chem. Phys. Lett. 2000, 321, 491–497. [Google Scholar] [CrossRef]
- Cheng, W.-D.; Xiang, K.-H.; Pandey, R.; Pernisz, U. C. Calculations of Linear and Nonlinear Optical Properties of H-silsesquioxanes. J. Phys. Chem. B 2000, 104, 6737–6742. [Google Scholar]
- Levitus, M.; Schmieder, K.; Ricks, H.; Shimizu, K. D.; Bunz, U. H. F.; Garcia-Garibay, M. A. Steps to Demarcate the Effects of Chromophore Aggregation and Planarization in Poly(phenyleneethynylene)s. 1. Rotationally Interrupted Conjugation in the Excited States of 1,4-Bis(phenylethynyl)benzene. J. Am. Chem. Soc 2001, 123, 4259–4265. [Google Scholar]
- Levitus, M.; Zepeda, G.; Dang, H.; Godinez, C.; Khuong, T.-A. V.; Schmieder, K.; Garcia-Garibay, M. A. Steps to Demarcate the Effects of Chromophore Aggregation and Planarization in Poly(phenyleneethynylene)s. 2. The Photophysics of 1,4-Diethynyl-2-fluorobenzene in Solution and in Crystals. J. Org. Chem. 2001, 66, 3188–3195. [Google Scholar]
- berg, K.; Berglund, A.; Edlund, U.; Eliasson, B. Prediction of Nonlinear Optical Responses of Organic Compounds. J. Chem. Inf. Comput. Sci. 2001, 41, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Halls, J. J. M.; Friend, R. H. Clean Electricity from Photovoltaics; Archer, M. D., Hill, R. D., Eds.; Imperial College: London, 2001; pp. 377–445. [Google Scholar]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Interacting Induced Dipoles Polarization Model for Molecular Polarizabilities. Application to Benzothiazole (A)-Benzobisthiazole (B) Oligomers: A-B13-A. J. Mol. Struct. (Theochem) 1998, 426, 105–116. [Google Scholar]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Torsional Effects on the Molecular Polarizabilities of the Benzothiazole (A)-Benzobisthiazole (B) Oligomer A-B13-A. J. Mol. Graphics 1996, 14, 245–259. [Google Scholar]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Polarization by the Effect of a Small Torsional Change in the Benzothiazole (A)-Benzobisthiazole (B) Oligomer A-B13-A. Molecules 1999, 4, 28–51. [Google Scholar]
- Torrens, F. A New Topological Index to Elucidate Apolar Hydrocarbons. J. Comput.-Aided Mol. Design 2001, 15, 709–719. [Google Scholar]
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. Molecules 2003, 8, 169–185. [Google Scholar]
- Torrens, F. A New Chemical Index Inspired by Biological Plastic Evolution. Indian J. Chem., Sect. A 2003, 42, 1258–1263. [Google Scholar]
- Torrens, F. A Chemical Index Inspired by Biological Plastic Evolution: Valence-Isoelectronic Series of Aromatics. J. Chem. Inf. Comput. Sci. in press.
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. J. Mol. Struct. (Theochem) 2003, 621, 37–42. [Google Scholar]
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. Mol. Diversity, in press.
- Randic, M. On Characterization of Molecular Branching. J. Am. Chem. Soc. 1975, 97, 6609–6615. [Google Scholar] [CrossRef]
- Hosoya, H. Topological Index. A Newly Proposed Quantity Characterizing the Topological Nature of Structural Isomers of Saturated Hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar]
- Gálvez, J.; García, R.; Salabert, M. T.; Soler, R. Charge Indexes. New Topological Descriptors. J. Chem. Inf. Comput. Sci. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- Kier, L. B.; Hall, H. L. Molecular Connectivity VII: Specific Treatment of Heteroatoms. J. Pharm. Sci. 1976, 65, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. Pharmacokinetic Optimization in Drug Research ; Testa, B., van de Waterbeemd, H., Folkers, G., Guy, R., Eds.; Helvetica Chimica Acta: Zurich, 2001; pp. 513–524. [Google Scholar]
- McClellan, A. L. Tables of Experimental Dipole Moments; Freeman: San Francisco, 1963. [Google Scholar]
- Tasi, G.; Mizukami, F. Scaled Effective One-Electron Method Based on G2 Theory: Results for Aliphatic Alkane Molecules. J. Chem. Inf. Comput. Sci. 1998, 38, 632–638. [Google Scholar] [CrossRef]
- Molina, V.; Smith, B. R.; Merchán, M. A Theoretical Study of the Electronic Spectrum of Styrene. Chem. Phys. Lett. 1999, 309, 486–494. [Google Scholar] [CrossRef]
- Molina, V.; Merchán, M. Theoretical Analysis of the Electronic Spectra of Benzaldehyde. J. Phys. Chem. A 2001, 105, 3745–3751. [Google Scholar]
- Sample availability: Not applicable.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Torrens, F. Valence Topological Charge-Transfer Indices for Reflecting Polarity: Correction for Heteromolecules. Molecules 2005, 10, 334-345. https://doi.org/10.3390/10020334
AMA Style
Torrens F. Valence Topological Charge-Transfer Indices for Reflecting Polarity: Correction for Heteromolecules. Molecules. 2005; 10(2):334-345. https://doi.org/10.3390/10020334
Chicago/Turabian StyleTorrens, F. 2005. "Valence Topological Charge-Transfer Indices for Reflecting Polarity: Correction for Heteromolecules" Molecules 10, no. 2: 334-345. https://doi.org/10.3390/10020334