Experimental
General
1H-NMR (δ, ppm) spectra of samples were obtained from CDCl
3 solutions on a Varian Gemini 2000 spectrometer operating at 300 MHz (
1H) and 75 MHz (
13C) frequency with tetramethylsilane as internal standard. IR and UV-VIS spectra of soluble samples were measure as CHCl
3 solutions on Perkin Elmer 781 spectrometer resp. Hewlett Packard 781 spectrometer. Optical rotations were measured on Perkin-Elmer 241 polarimeter. Diffuse reflectance FTIR (DRIFTS) spectrum of modified silica gel was measured as a mixture of 5% sample and 95% KBr in an environmental chamber at 150°C under vacuum on a Bruker Equinox FTIR. The porosimetry data were measured on Micromeritics ASAP2100 system using nitrogen as adsorbate at 77K. Samples were degassed at 120°C until a constant vacuum of 5 microns Hg was obtained. Solvents were purified and dried according to standard published methods. (
R, p
S)-PPFNMe was prepared according to described methods [
16].
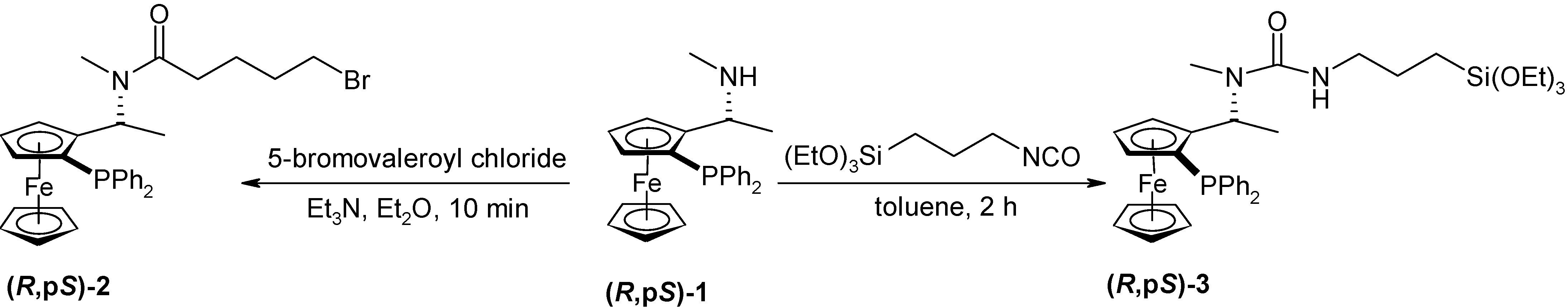
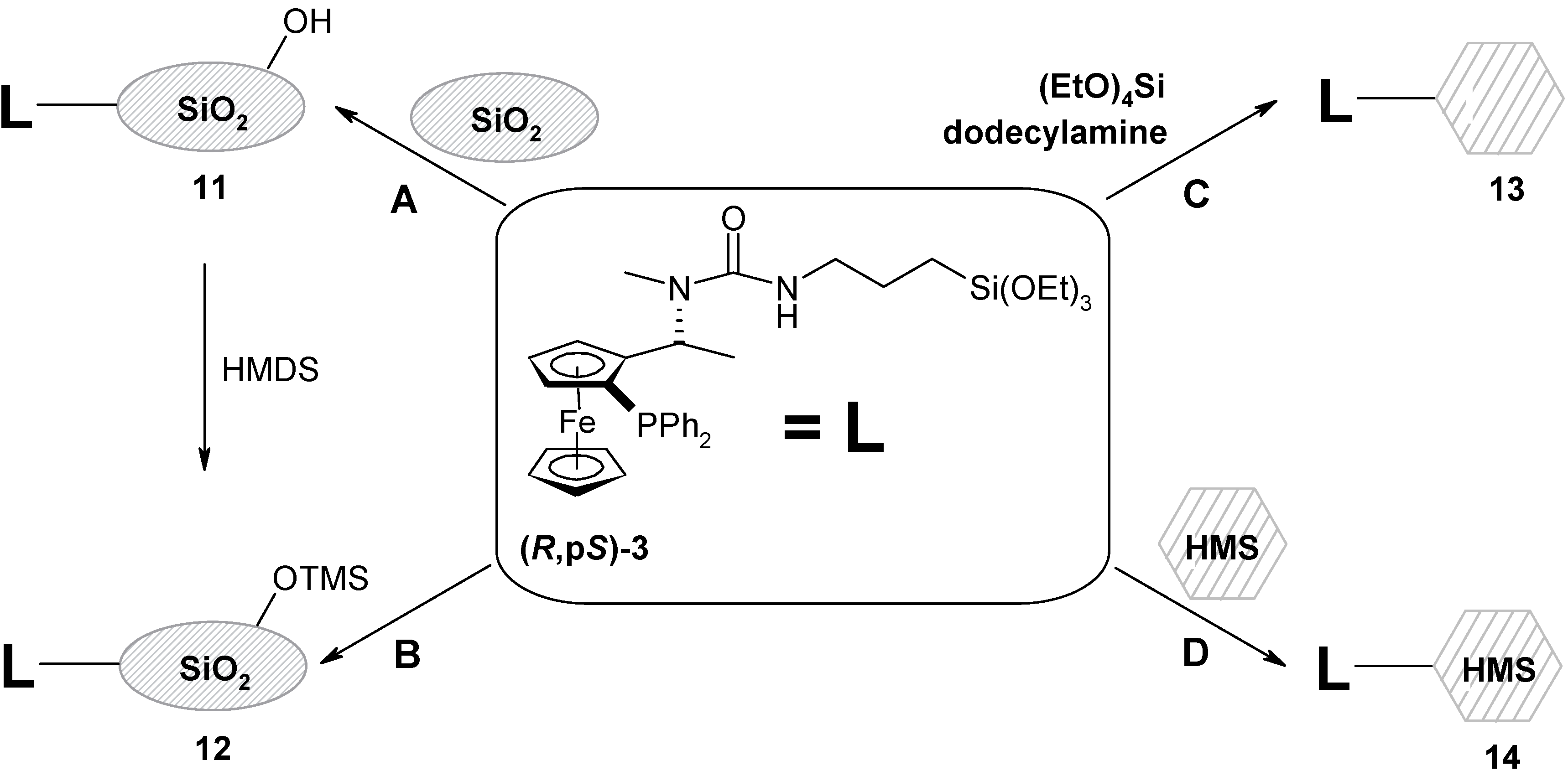
Preparation of (R,pS)-N-methyl-N-1-[2-(diphenylphosphanyl)ferrocenyl]ethyl-N´-[3-triethoxysilyl)-propyl]urea (R,pS)- 3:
(R,pS)-PPFNMe (818 mg, 1.91 mmol) stored under nitrogen was dissolved in dry toluene (16 mL) and ICPTES (498 μL, 2 mmol, 1.05 eq.) was added dropwise. The orange solution was stirred for 2 h at r.t. under nitrogen. The solvent was removed on a rotavapor and the residue was purified by the column chromatography (SiO2, 80 g, diethyl ether). The product was obtained as an orange semi-solid material (1.18 g, 93 %), [α]D21 = -254, [α]578 = -274, [α]546 = -356 (c = 0,37; chloroform). For C35H47O4N2PSiFe (674.68) calcd.: 62.31 %C; 7.02 %H; 4.15 %N; found.: 62.17 %C; 6.97 %H; 3.84 %N; TLC: RF = 0,51 (SiO2, Et2O); 1H-NMR δ: 0.55 (t, 2H, -CH2Si, 3JHH = 8.52 Hz); 1.23 (t, 9H, -OCH2CH3, 3JHH = 7.14 Hz); 1.38 – 1.48 (m, 2H, -CH2); 1.42 (d, 3H, -CH3, 3JHH = 6.87 Hz); 1.66 (bs, 1H, -NH); 1.99 (s, 3H, -NCH3); 2.85 (dt, 1H, -NCHA, 2JHH = 7.42 Hz, 3JHH = 12.91 Hz); 3.05 (dt, 1H, -NCHB, 2JHH = 7.42 Hz, 3JHH = 12.91 Hz); 3.55 (t, 1H, Fc, 3JHH = 5.22 Hz); 3.81 (q, 6H, -OCH2CH3, 3JHH = 6.87 Hz); 4.04 (s, 5H, cp); 4.30 (t, 1H, Fc, 3JHH = 2.47 Hz); 4.47 (bs, 1H, Fc); 5.73 (dq, 1H, -CH, 3JHH = 6.56 Hz, JHP = 1.92 Hz); 7.16-7.24 (m, 5H, -PPh2); 7.33-7.36 (m, 3H, -PPh2); 7.16-7.24 (m, 5H, -PPh2); 7.50-7.56 (m, 2H, -PPh2); 13C-NMR δ: 7.75 (-CH2Si); 16.02 (-CH3); 18.33 (-OCH2CH3) 23.47 (-CH2); 43.40 (-CH2N); 49.05 (d, -CH, 3JCP = 7.73 Hz); 58.35 (-OCH2); 6870 (-NCH3); 69.91 (cp); 70.13 (d, Fc, 2JCP = 4.01 Hz); 72.69 (d, Fc, 2JCP = 4.87 Hz); 76.79 (d, CFci, 3JCP = 10.31 Hz), 94.33 (d, -CFci, 2JCP = 24.91 Hz); 127.82; 127.91; 128.07, 128.16; 128.97; (-PPh2(m, p), -Ph), 133.41 (d, -PPh2(o), 2JCP = 19.75 Hz); 135.56 (d, -PPh2(o), 2JCP = 20.90 Hz); 137.44 (d, -PPh2(i), 1JCP = 9.73 Hz); 139.71 (d, -PPh2(i), 1JCP = 10.88 Hz); 157.32 (CO); UV VIS: λmax (nm) 210, 248; IR: υ (cm-1) 1420 (CCFc); 1620 (CO) (CHCl3).
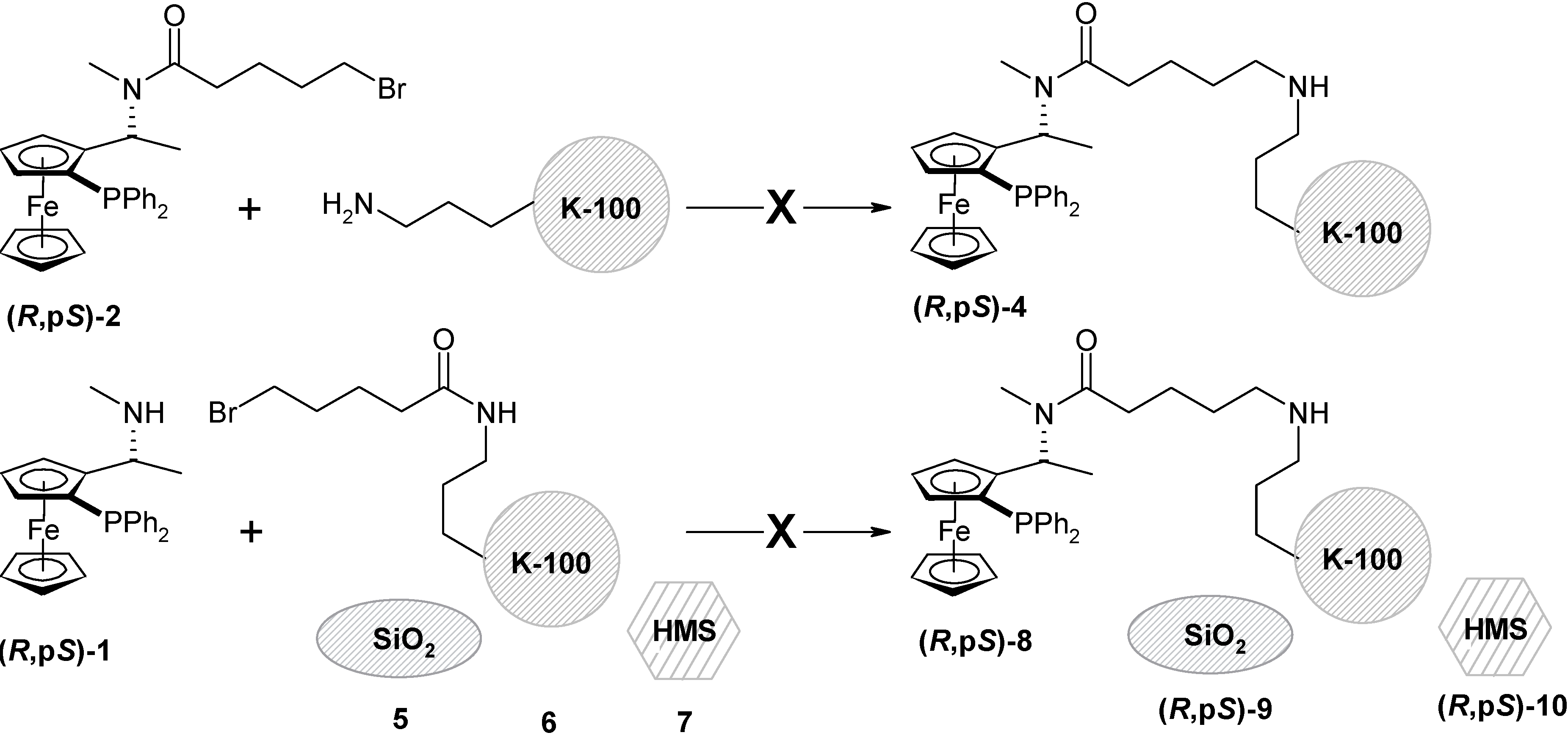
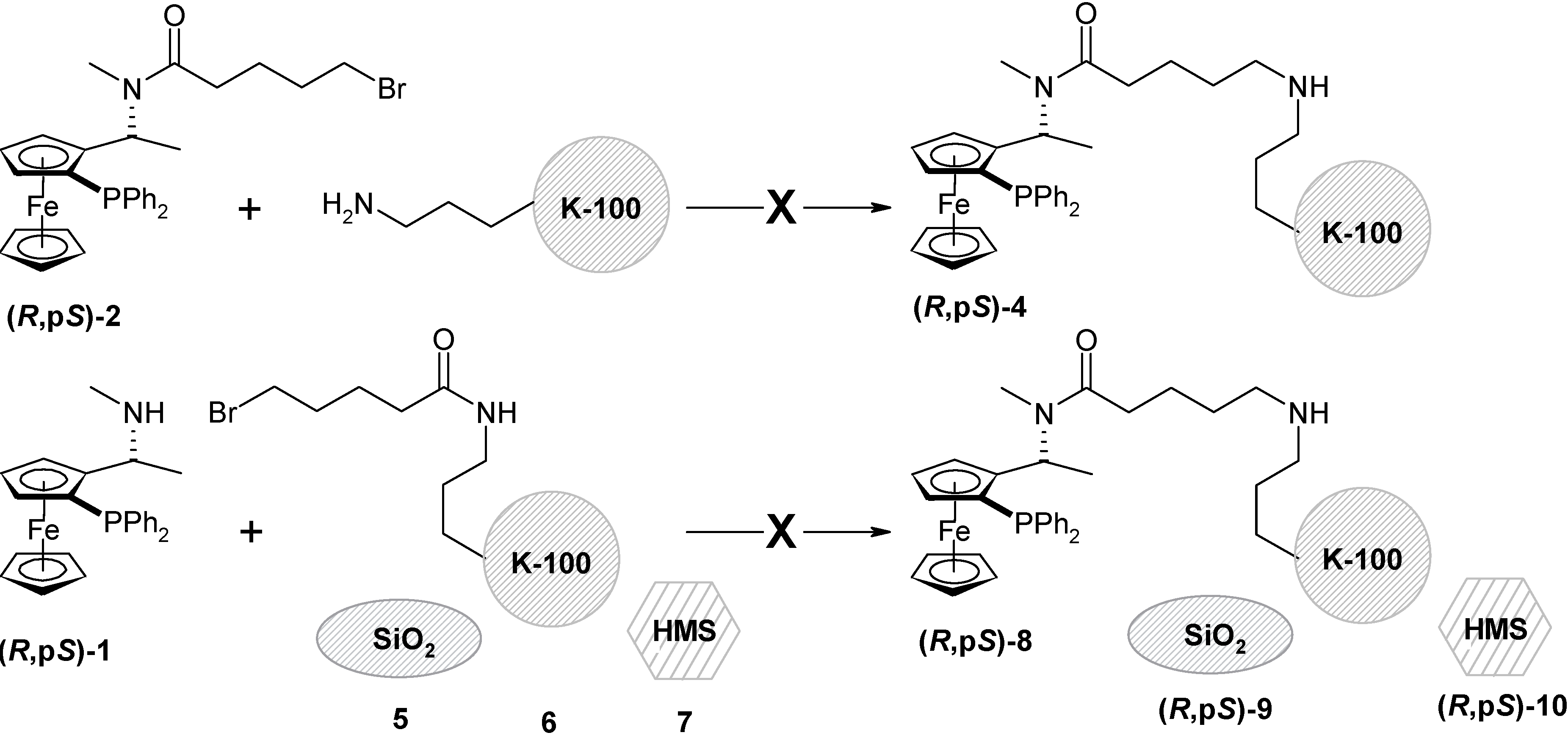
The immobilization of (R,pS)-3 to the amorphous silica gel, preparation of (R,pS)-11
Silica gel (5 g) was dried (4 h, 150 °C, 1 mm Hg) and stored under argon. Dry toluene was added (15 mL) and it was stirred for 30 min. at r.t. The ligand (R,pS)-3 (675 mg; 1 mmol) was dissolved in dry toluene (20 mL) and added to the suspension of the silica gel. The mixture was stirred at 70 °C for 4 h and then for 16 h at r.t. under argon. The silica gel was filtered off, washed with diethyl ether, dichloromethane, acetone and ethanol and dried (3 h, r.t., 1 mm Hg). The modified silica gel (R,pS)-11 was obtained as an yellow powder (4.9 g). Specific surface: 467 m2/g; pore volume: 0.76 cm3/g; average pore diameter: 63 Å (broad size distribution), C = 69.0 (polar surface); DRIFTS spectra: υ (cm–1) 970 (-SiOdef), 1438 (C-Hdef), 1536, 1560 (aryl), 1625, 1523 (urea, SiOH), 2850, 2913, 2979 (aliph.), 3064 (aryl C-H), 3735 (isolated silanol groups); Thermal analysis: weight loss at 150°C (3.1% - water and solvents); at 180 – 250°C (0.5%); 260 – 630°C (3%; organic material). AAS analysis (Fe): 51μmol/g.
The protection of the silanol groups of the modified silica gel (R,pS)-11: preparation of (R,pS)-12
Modified silica gel (R,pS)-11 (2,5 g) was suspended in dry toluene (20 mL), HMDS was added in one portion (2.5 mL; 12 mmol) and the resulting mixture was refluxed for 1 h under argon. Then it was cooled down, the silica gel was filtered off, washed with diethyl ether, dichloromethane, acetone and ethanol and dried (3 h, r.t., 1 mm Hg). Modified silica gel (R,pS)-12 was obtained as a yellow powder (2.5 g). Surface area: 347 m2/g; pore volume: 0.46 cm3/g; average pore diameter: 53 Å (broad size distribution), C = 16.2 (decrease of the surface polarity); DRIFTS spectra: υ (cm–1) 970 (-SiOdef), 1438 (C-Hdef), 1536, 1560 (aryl), 1625, 1523 (urea, SiOH), 2850, 2913, 2979 (aliph.), 2846, 2902, 2960 (CH3), 3064, 3100 (aryl C-H); Thermal analysis: weight loss at 150°C (0.4% - water and solvents); 180 – 350°C (1.6%); 350 – 750°C (4.4%; organic material). AAS analysis (Fe): 50 μmol/g.
Sol-gel-template immobilization of (R,pS)-3: preparation of (R,pS)-13
Dodecylamine (357 mg, 1.94 mmol) was dissolved in the mixture of ethanol (3.3 mL) and distilled water (3.8 mL). The mixture was stirred for 20 min. at r.t. and bubbled through with nitrogen to form a clear colorless solution. It was then added to the (R,pS)-3 (487 mg, 0.71 mmol) together with tetraethoxysilane (1.43 mL, 6.37 mmol, 9 eq. with regards to (R,pS)-3)). The mixture was stirred for 20 h under nitrogen. A white-yellow thick mixture was formed. The solid was filtered off and the template was removed by continual extraction in a Soxhlet extractor for 7 h with degassed ethanol (500 mL) under nitrogen. Modified HMS (R,pS)-13 was obtained as yellow powder (800 mg). Surface area: 407 m2/g; pore volume: 0.17 cm3/g (pores formed by the SGT mechanism); 0.175 cm3/g (pore not formed by SGT mechanism); average pore diameter: 14 Å. DRIFTS spectra: υ (cm–1) 968 (-SiOdef), 1434, 1477 (C-Hdef), 1519, 1635, 1644 (urea, SiOH), 1536, 1560 (aryl), 2850, 2898, 2908, 2933, 2977 (aliph.), 2846, 2902, 2960 (CH3), 3056, 3066, 3095 (aryl C-H), 3731, 3644, 3411 (isolated silanol groups); Thermal analysis: weight loss at 180°C (4,4% - water and solvents); at 180 – 290°C (4.7%); 290 – 750°C (17.3%; organic material). 13C-CPMAS (ppm) 133-145 (aryl-P); 78 (Cp); 57 (N-CH3); 51 (N-CH2); 32 and 36 (CH2CH2CH2 and CH-Cp); 24 (CH3-CH); 17 (CH2-Si); AAS analysis (Fe): 478 μmol / g.
The immobilization of (R,pS)-3 to the HMS: preparation of (R,pS)-14
HMS (3 g) was dried (4 h, 150 °C, 1 mm Hg) and stored under argon. Dry toluene (30 mL) was added and it was stirred for 30 min. at r.t. The ligand (R,pS)-3 (256 mg; 0.6 mmol) was dissolved in dry toluene (10 mL) and added to the silica gel suspension. The mixture was stirred at 70 °C for 4 h and then for 16 h at r.t. under argon. The silica gel was filtered off, washed with diethyl ether, dichloromethane, acetone and ethanol and dried (3 h, r.t., 1 mm Hg). The modified silica gel (R,pS)-14 was obtained as an pale yellow powder (3.1 g); Surface area: 1092 m2/g; pore volume: 1.022 cm3/g; average pore diameter: 25.1 Å; DRIFTS spectra: υ (cm–1) 1436, 1475 (aliphatic C-Hdef), 1540, 1558 (aryl C-Hdef), 1525, 1636, 1659 (urea), 2854, 2902, 2936, 2984 (C-Hstr), 3106, 3061 (aryl C-Hstr), 3535 (H-bonded silanol) 3739 (isolated silanol); Thermal analysis: weight loss at 150°C (2.6% - water and solvent); at 215 – 450°C (gradual loss); 450 – 645°C (6.2 % in total); 650 - 800°C (1.1 %); AAS analysis (Fe): 120μmol/g.
Preparation of (R,pS)-N-phenyl-N´-methyl-N´-1-[2-(diphenylphosphanyl)ferrocenyl]ethylurea (R,pS)-20
(R,pS)-PPFNMe (100 mg; 0.23 mmol) was dissolved in dry toluene (3 mL) and ICPTES (28 μL; 0.26 mmol; 1.1 eq.) was added dropwise. The orange solution was stirred for 2 h at r.t. under argon. The solvent was removed and the residue was purified by the column chromatography (SiO2, 15 g, diethyl ether). The product was obtained as an orange semi-solid substance (124 mg, 99 %), t.t. = 91 – 93°C. For C32H31N2OPFe (546.24) calcd.: 70.36 %C; 5.72 %H; 5.13 %N; found.: 69.65 %C; 5.54 %H; 4.88 %N; TLC: RF = 0.52 (SiO2, Et2O); 1H-NMR δ: 1.51 (d, 3H, -CH3, 3JHH = 6.96 Hz); 2.14 (s, 3H, -NCH3); 2.17 (s, 1H, -NH); 3.86 (t, 1H, Fc, 3JHH = 2.19 Hz); 4.07 (s, 5H, cp); 4.35 (t, 1H, Fc, 3JHH = 2.38 Hz); 4.51 (t, 1H, Fc, 3JHH = 2.56 Hz); 5.77 (q, 1H, -CH, 3JHH = 6.78 Hz); 6.01-7.57 (14H, PPh2, -Ph); 13C-NMR δ: 16.39 (-CH3); 28.53 (-NCH3); 49.95 (d, -CH, 3JCP = 6.50 Hz); 69.09; 70.30; 70.34; 72.45 (d, 2JCP = 4.47 Hz), (-Fc); 70.22 (cp); 93.66 (d, -Ccpi, 2JCP = 24.78 Hz); 119.29; 122.35; 128.06; 128.18; 128.30; 128.36; 128.41; 128.76; 129.14 (-PPh2(m, p), -Ph), 133.42 (d, -PPh2(o), 2JCP = 19.19 Hz); 133.97 (d, -PPh2(o), 2JCP = 21.13 Hz); 136.89 (d, -PPh2(i), 1JCP = 8.53 Hz); 139.11 (d, -PPh2(i), 1JCP = 9.75 Hz); 139.68 (-Ci, -Ph);; 154.49 (CO); UV VIS: λmax (nm) 208, 244; IR: υ (cm-1) 1415 (CCFc); 1640 (CO) (CHCl3).
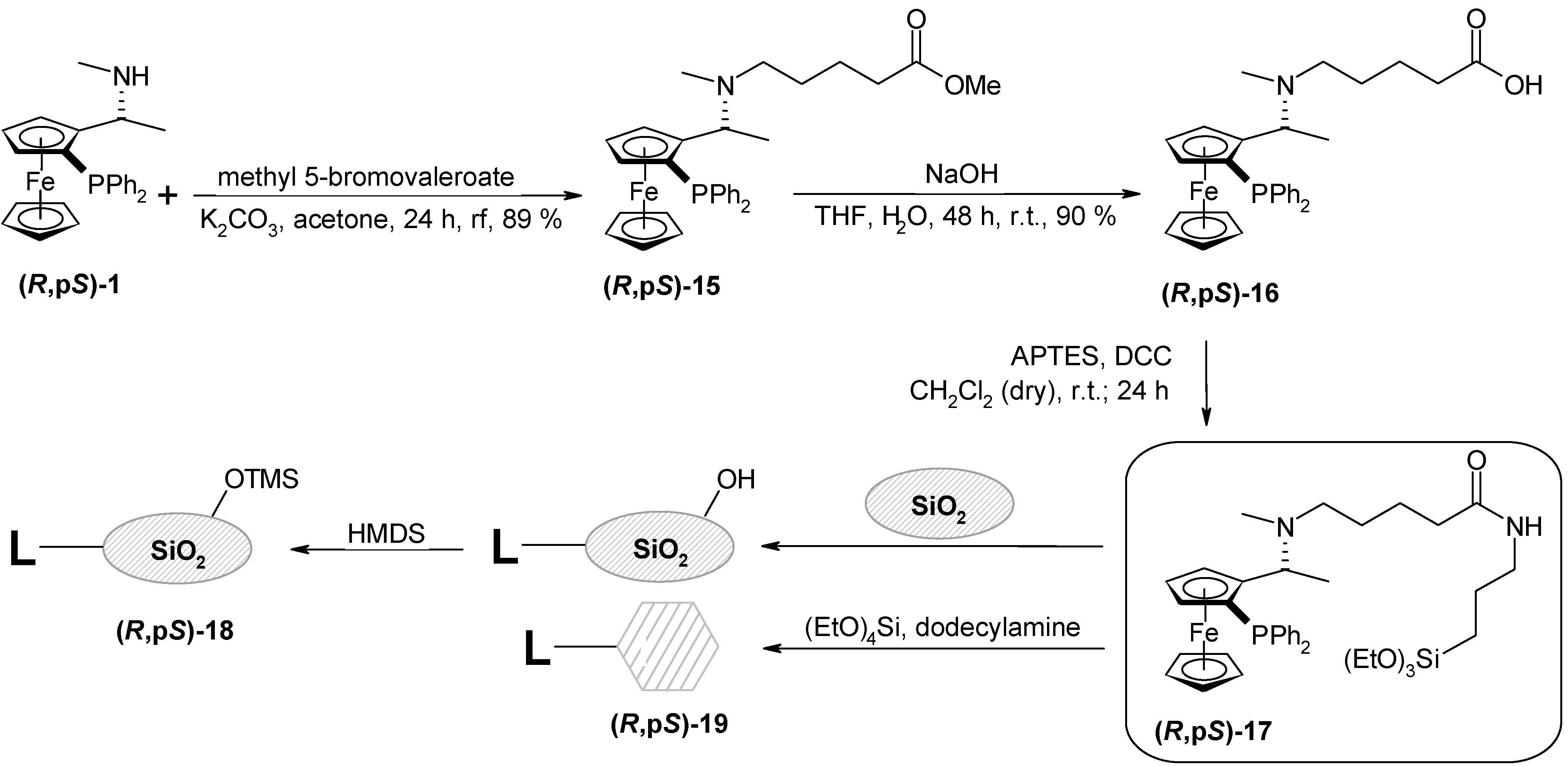
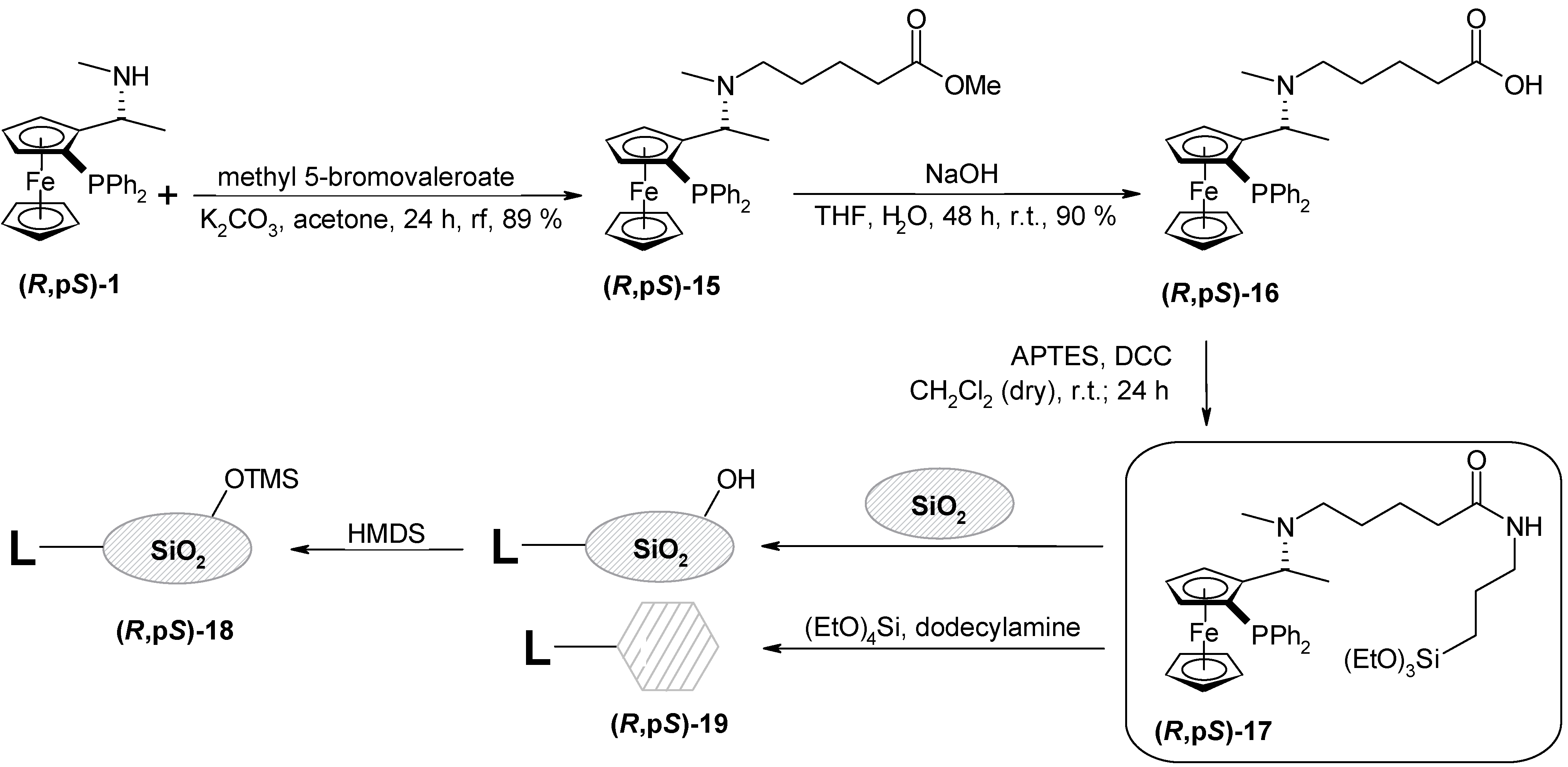
Preparation of methyl (R,pS)-5-{N-methyl-1-[2-diphenylphosphanyl)ferrocenyl]ethylamino}valerate (R,pS)-15
(R,pS)-1 (1.12 g; 2.62 mmol), and potassium carbonate (543 mg; 3.93 mmol, 1.5 eq.) were mixed in acetone (5 mL). Methyl 5-bromovalerate (413 μL; 2,88 mmol; 1,2 ekv.) was added and the resulting mixture was refluxed under nitrogen for 23 h. After cooling down, the solvent was removed on RV, water (80 ml) was added to the residue and it was extracted with dichloromethane (3x30 mL). Combined organic layers were dried over Na2SO4 and the solvent was removed. The product was isolated by the column chromatography on SiO2 (30 g) hexane-ethyl acetate mixture (1:1) as an eluent. The product (R,pS)-15 was obtained from the first fraction as an orange oil (1.26 g, 89 %); [α]D21 = -155, [α]578 = -167, [α]546 = -214 (c = 0.29; chloroform). For C31H36O2NPFe (541.45) calcd.:68.76 %C; 6.70 %H; 2.59 %N; found. 70.69 %C; 8.02 %H; 2.30 %N; 1H-NMR (CDCl3): δ (ppm) 0.78-0.89 (m, 2H, -CH2); 1.17-1.27 (m, 2H, -CH2); 1.25 (d, 3H, -CH3, 3JHH = 6.59 Hz); 1.69 (s, 3H, -NCH3); 2.05 (m, 2H, -CH2CO); 2.11 – 2.18 (m, 1H, -CHAN); 2.25 – 2.34 (m, 1H, -CHBN); 3.63 (s, 3H, -OCH3); 3.84 (m, w1/2 = 2.20 Hz; 1H, -Fc); 3.92 (s, 5H, cp); 4.22 – 4.26 (m, 2H, -Fc, -CH); 4.38 (dd, 1H, Fc, 3JHH = 2.20 Hz; 3JHH = 3.58 Hz); 7.08 – 7.59 (m, 10H, -PPh2); 13C-NMR (CDCl3): δ (ppm) 9.18 (-CH3); 22.64; 27.11 (-CH2); 29.71 (-NCH3); 33.97 (-CH2CO); 51.32 (-OCH3); 53.68 (-NCH2); 57.50 (d, -CH, 3JCP = 5.15 Hz); 68.37; 71.78; 71.90 (Fc); 69.58 (cp); 76.12 (d, -Fc, 3JCP = 9.17 Hz); 97.29 (d, CFci, 2JCP = 22.30 Hz); 128.73; 127.27 (d, 3JCP = 6.30 Hz); 127.79 (d, 3JCP = 7.73 Hz, -PPh2); 132.30 (d, -PPh2(o), 2JCP = 18.04 Hz); 135.42 (d, -PPh2(o), 2JCP = 21.47 Hz); 138.91, 141.35 (-PPh2(i)); 174.22 (-CO); UV VIS: λmax (nm) 210, 224; IR: υ (cm-1) 1420 (CCFc); 1710 (CO) (CHCl3).
Preparation of (R,pS)-5-{N-methyl-1-[2-diphenylphosphanyl)ferrocenyl]ethylamino}valeric acid (R,pS)-16
(R,pS)-15 (1.26 g; 2,33 mmol) was dissolved in THF (17 mL) and the solution of sodium hydroxide (1.86 g; 46.5 mmol; 20 eq.) in distilled water (3 mL) was added. The mixture was stirred at r.t. under nitrogen for 48 h. The solvent was removed and water (15 mL) followed by 20 % HCl (30 mL) was added. Then it was extracted with dichloromethane (3x20 mL). Combined organic layers were dried over Na2SO4 and removed from the solvent. The yellow powder (hydrochloride) was stirred for 10 min. with 13% aqueous NH3 (20 mL) and it was extracted with dichloromethane (2x20 mL). The organic layer was dried over Na2SO4 and removed from the solvent. Product was purified by the column chromatography on SiO2 (5 g) with methanol as an eluent. The product (R,pS)-16 was obtained as an yellow solid (1.1 g, 90 %); t.t. = 65 – 68°C; [α]D21 = -311, [α]578 = -341, [α]546 = -452 (c = 0.45, chloroform). For C30H34O2NPFe (527.41) calcd.: 68.32 %C; 6.49 %H; 2.65 %N; found.: 64.07 %C; 6.63 %H; 2.32 %N; TLC: RF = 0.51 (SiO2, Et2O); 1H-NMR (CDCl3): δ 0.94 – 1.19 (m, 4H, -CH2CH2); 1.46 (d, 3H, -CH3, 3JHH = 6.59 Hz); 1.85 (s, 3H, -NCH3); 1.92-2.01 (m, 2H, -COCH2); 2.17–2.24 (m, 2H, -CH2N); 3.88 (s, 5H, cp); 4.02 (m, w1/2 = 5.37 Hz, 1H, -Fc); 4.32 – 4.40 (m, 2H, -Fc, -CH); 4.45 (m, w1/2 = 5.86 Hz, 1H, Fc); 7.14-7.61 (m, 10H, -PPh2); 13C-NMR (CDCl3): δ 18.44 (-CH3); 23.27; 26.16 (-CH2); 34.44 (-NCH3); 36.42 (-CH2CO); 53.82 (-NCH2); 57.67 (d, -CH, 3JCP = 8.59 Hz); 69.46; 70.08; 72.07 (d, 3JCP = 5.15 Hz); 76.19 (d, 3JCP = 11.17 Hz) (Fc); 69.84 (cp); 94.44 (CFci); 127.56; 127.74 (d, 3JCP = 6.30 Hz); 127.96 (d, 3JCP = 8.01 Hz); 128.98 (-PPh2); 132.36 (d, -PPh2(o), 2JCP = 18.32 Hz); 135.11 (d, -PPh2(o), 2JCP = 22.33 Hz); 138.60; 140.25 (d, -PPh2(i), 2JCP = 7.73 Hz); 179.89 (-CO); UV VIS: λmax (nm) 210, 246; IR: υ (cm-1) 1420 (CCFc); 1700 (CO) (CHCl3).
Condensation of APTES with (R,pS)-5-{N-methyl-1-[2-diphenylphosphanyl)ferrocenyl]ethyl-amino}-valeric acid – preparation of (R,pS)-17
(R,pS)-16 (800 mg; 1.52 mmol) and DCC (328 mg; 1.59 mmol) were dissolved in dry dichloromethane (8 ml). APTES (372 μL; 1.59 mmol; 1.05 eq.) was added and the resulting orange mixture was stirred at r.t. for 24 h under argon. Diethyl ether (20 mL) was added and the insoluble N,N´-dicyclohexylurea was filtered off. Finally the solvent was removed on RV. An orange oil (980 mg) was obtained and used without further purification.
Immobilization of (R,pS)-17 to the amorphous silica gel, preparation of (R,pS)-18
Done by the same procedure as in the case of (R,pS)-11. The yellow powder (1.98 g) was subsequently submitted for the protection of free silanol groups by the procedure described for the (R,pS)-11. Yellow powder (R,pS)-18 was obtained (1.86 g); Surface area: 373 m2/g; pore volume: 0.53 cm3/g; average pore diameter: 55 Å; DRIFTS spectra: υ (cm–1) 1410, 1436 (aliphatic C-Hdef), 1518, 1636, 1648 (amide), 2843, 2902, 2961 (aliphatic C-Hstr), 3065, 3102 (aryl C-Hstr), 3502, 3687 (H-bonded silanol); Thermal analysis: weight loss at 150°C (0.5% - water); at 200 - 800°C (5.7 % - organics + TMS); AAS analysis (Fe): 44 μmol/g.
Immobilization of (R,pS)-17 by SGT method, preparation of (R,pS)-19
Done by the same procedure as in the case of (R,pS)-13. Modified HMS (R,pS)-19 was obtained as a pale yellow powder (311 mg). AAS analysis (Fe): 50 μmol/g
Pd(0)-catalyzed allylic substitution of rac-E-1,3-diphenyl-3-acetoxyprop-1-ene
Pd2(dba)3.CHCl3 (1.29 mg; 1.25 μmol; 1 mol. % Pd), ligand (2.75 μmol; 2 eq. to Pd) and the substrate (63 mg; 0.25 mmol) were suspended in dry THF (2 mL) under argon and were stirred for 20 min. at r.t.. Sodium hydride (7.8 mg; 0.33 mmol; 1.3 eq.) was suspended in another flask in dry THF (2 ml) and dimethylmalonate was slowly added (42.5 μL; 0.37 mmol; 1.5 eq.). The solution was added to the first solution and the reaction mixture was stirred at 40 °C. After the reaction, the silica gel was filtered off, water (10 ml) was added to the filtrate and it was extracted with diethyl ether (2x20 mL). Combined organic layers were dried with Na2SO4 and the solvent was removed on RV. The product was isolated by the column chromatography (SiO2, 10 g, Hex -EA = 7:1). The product was isolated as clear colorless oil; TLC: RF = 0.54 (SiO2, Hex:EA = 3:1); 1H-NMR (CDCl3): δ 3.51 (s, 3H, -COOCH3), 3.70 (s, 3H, -COOCH3), 3.95 (d, 1H, H1, 3JHH=10.80 Hz), 4.27 (dd, 1H, H2, 3JHH=8.42 Hz; 3JHH=10.98 Hz), 6.32 (dd, 1H, H3, 3JHH=8.60 Hz; 3JHH=15.75 Hz); 6.48 (d, 1H, H4, 3JHH=15.75 Hz), 7.17 – 7.34 (m, 10H, -PPh2); the enantiomeric purity was determined by 1H-NMR with Eu(hfc)3 (0,3 eqv.) as a chiral shift agent.
Rh-catalyzed hydrogenation of (Z)-α-acetamidocinnamic acid
[RhCl(C6H10)]2 (2.8 mg, 6.2 μmol, 1 mol. % Rh), ferrocenyl ligand (6.3 μmol) and the substrate (257 mg, 1.25 mmol) were placed in a Teflon reactor under nitrogen. Degassed methanol (8 mL) was added. The reactor was placed in an autoclave, hydrogen was then introduced after three successive substitutions of nitrogen with hydrogen (50 atm.). Reactions were performed at r.t. for an appropriate time. The catalyst was filtered off, washed with methanol, dried (r.t., 1 mm Hg, 2 h and stored under argon for next run. The filtrate was removed from the solvent. The yield was calculated according to the 1H-NMR spectrum (d6-DMSO) of a crude sample. 1H-NMR (d6-DMSO): δ 1.77 (s, 3H, COCH3); 2.83 (dd, 2JAB = 13.7 Hz, 3JHH = 9.3 Hz, 1H, CHA), 3.04 (dd, 2JAB = 13.7 Hz, 3JHH = 4.4 Hz, 1H, CHB); 4.38 (m, 1H, -CH); 7.16-7.30 (m, 5H, Ph); 8.12 (d, 3JHH = 8.2 Hz, NH).


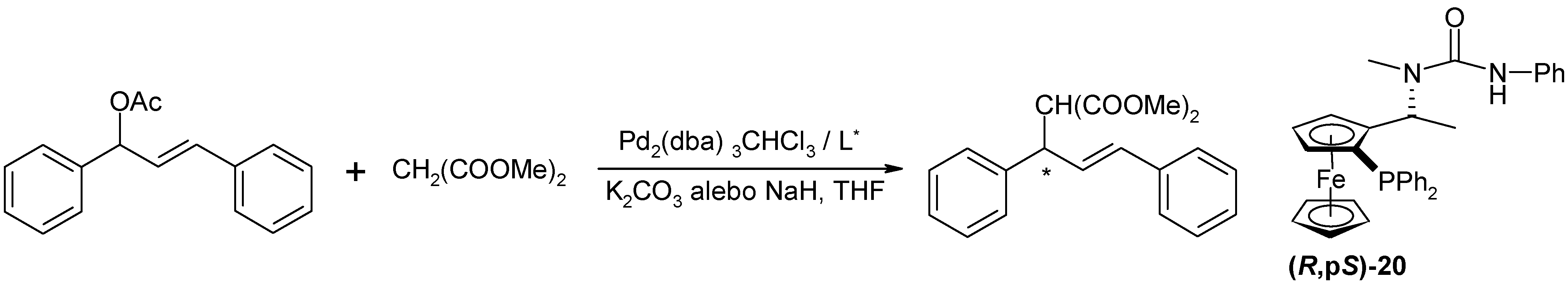

{kind=link}
{kind=link}
{kind=link}
{kind=link}
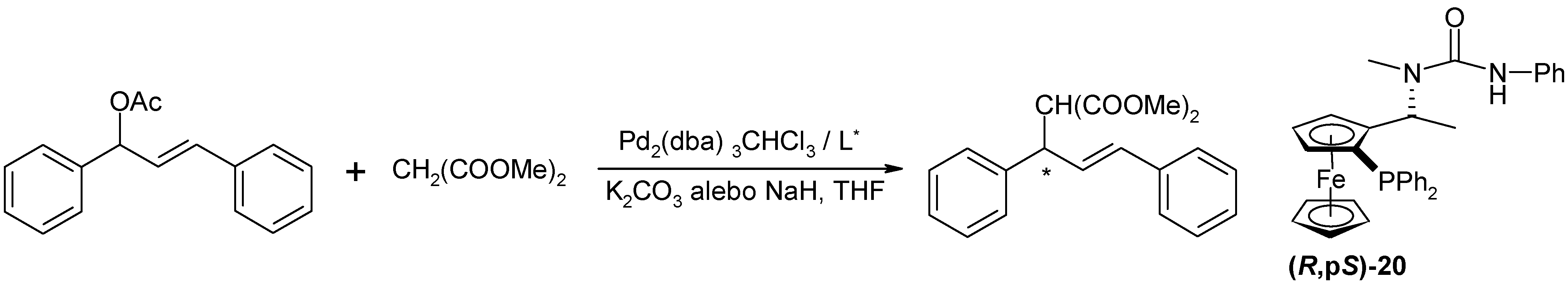{kind=link}
{kind=link}
