Pyrimidine Acyclo-C-Nucleosides by Ring Transformations of 2-Formyl-L-arabinal

1
Institut für Chemie der Universität Rostock, D-18051 Rostock, Germany
2
Leibniz-Institut für Organische Katalyse, Buchbinderstr. 5–6, D-18055 Rostock, Germany
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(8), 837-842; https://doi.org/10.3390/10080837
Submission received: 29 September 2004
/
Revised: 5 April 2005
/
Accepted: 5 April 2005
/
Published: 31 August 2005
{kind=link}
{kind=link}
Abstract
:The protected 2-formyl-L-arabinal 2 reacted with thiourea and cyanamide in the presence of sodium hydride to afford via ring transformations the 5-[1R,2S-1,2-bis(benzyloxy)-3-hydroxypropyl]-1,2-dihydropyrimidines 3 and 4, respectively. Similarly, treatment of 2 with 3-amino-2H-1,2,4-triazole yielded 6-[1R,2S-1,2-bis(benzyloxy)-3-hydroxypropyl][1,2,4]-triazolo[1,5-a]pyrimidine (5).
Introduction
Because of their push-pull activated carbon-carbon double bond [1,2,3], 2-formylglycals [4,5,6] synthesized by a Vilsmeier-Haack reaction of O-benzyl protected hexose glycals are a versatile class of compounds which allowed a nucleophilic attack of dinucleophiles at C-1 under ring opening of the glycals followed by recyclization involving the formyl group to give acyclo-C-nucleoside analogues [6,7,8,9,10,11,12]. The protected 2-formyl-L-arabinal 2 was easily prepared from O-benzylated L-arabinal (1) by treatment with phosphoryl chloride and N,N-dimethylformamide [12]. In this paper we report the ring transformation reactions of 2-formyl-L-arabinal 2 with some nitrogen nucleophiles to obtain pyrimidine C-nucleoside analogue and related compounds.
Results and Discussion
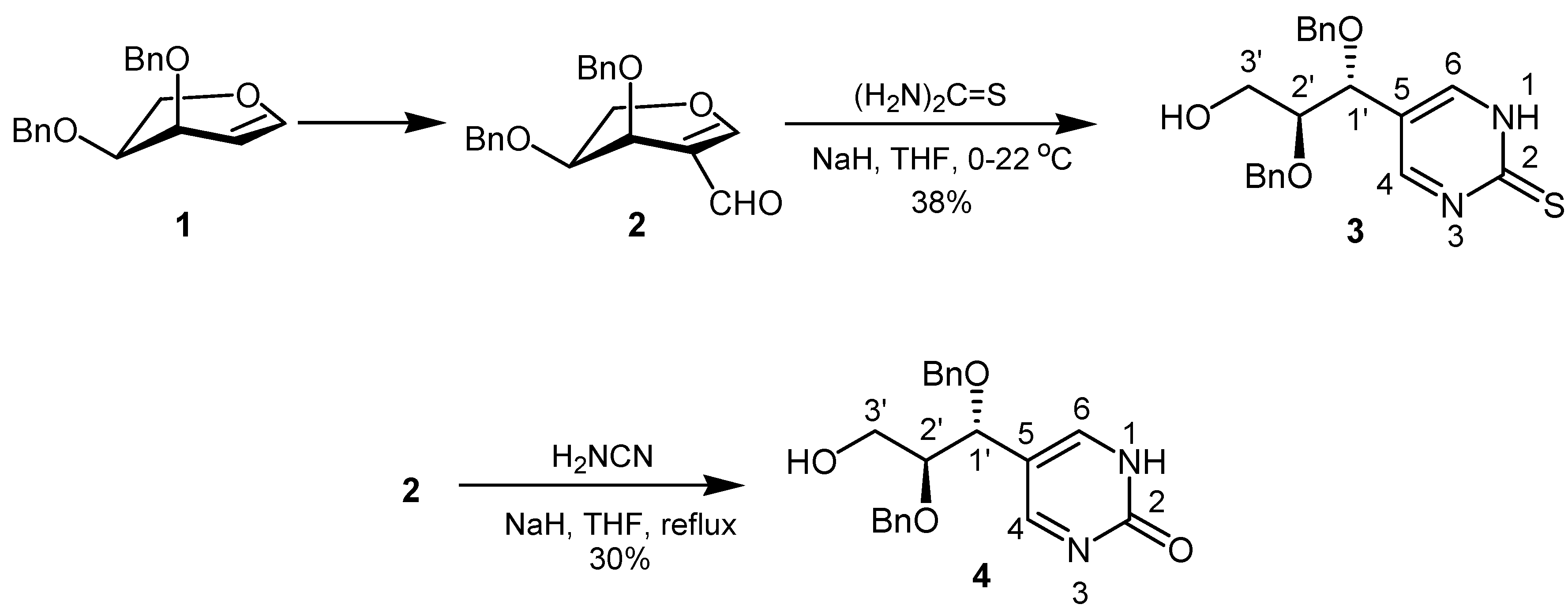
Thiourea was reacted with 2-formyl-L-arabinal 2 in the presence of sodium hydride in tetrahydrofuran to afford through the sequential combination of addition-elimination and ring closure reaction the required pyrimidine C-nucleoside analogue 3 in 38% yield as a pale yellow syrup (Scheme 1).
Scheme 1.
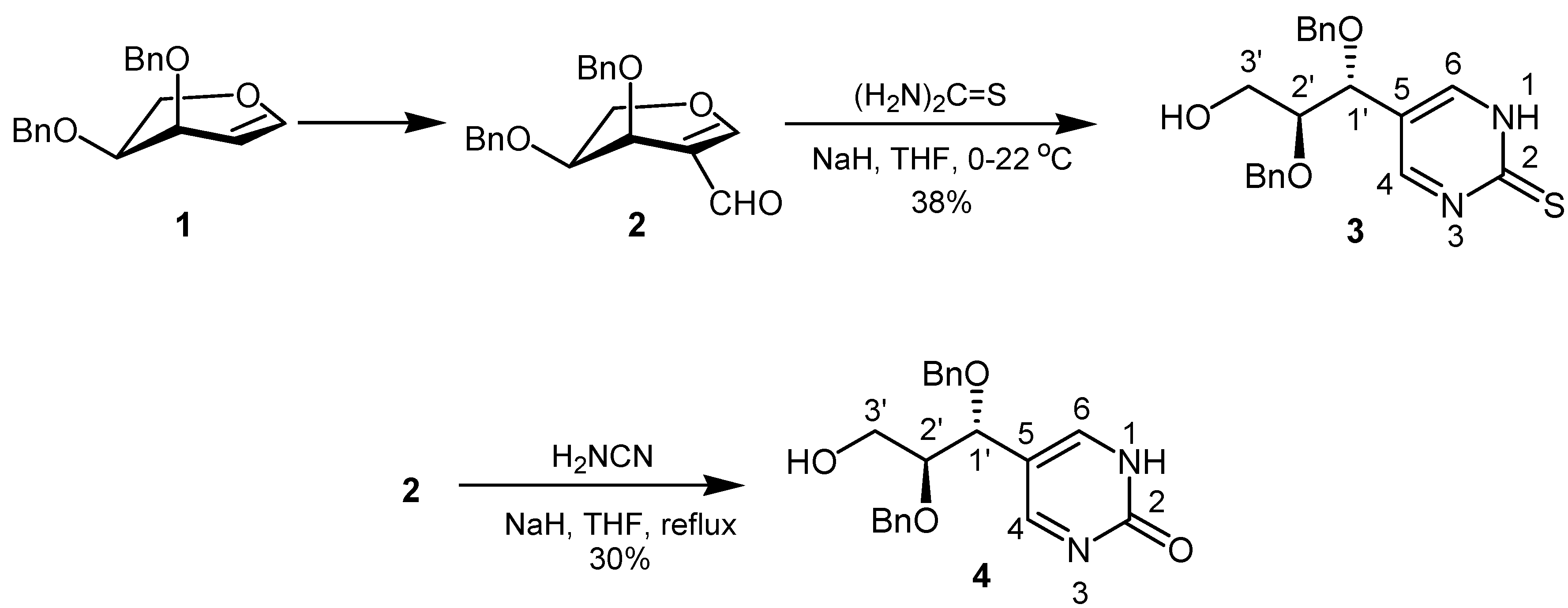
Compound 3 was characterized by 1H- and 13C-NMR as well as IR and mass spectroscopy. In the 1H-NMR spectrum, H-4 and H-6 exhibit only one signal at δ = 8.50, which is due to the tautomerization involving the proton on the N atoms. In the 13C-NMR spectrum C-4 and C-6 appeared also as one signal at δ = 157.0. For C-2 a chemical shift of δ = 170.7 was found. The IR spectrum showed a typical absorption for the associated OH group at 3418 cm–1. Furthermore, the mass spectrum confirmed the presence of the thiourea structural element by giving a [M+H]+ peak at 383.
In order to prepare the corresponding 5-[1R,2S-1,2-bis(benzyloxy)-3-hydroxy-propyl]-1,2-dihydro-pyrimidin-2-one (4), formylarabinal 2 was treated under reflux with an excess of cyanamide in tetrahydrofuran for 30 hours in the presence of sodium hydride. As first step in this reaction we assume a nucleophilic attack of the cyanamide amino group at formyl carbon followed by hydrolysis of the nitrile to give the carboxamide group. Ring transformation to 4 then occurred through carboxamide attack at C-1.
The carbonyl resonance was found at δ = 162.8 in the 13C-NMR spectrum, while H-4 and H-6 appeared as a common singlet at δ = 8.25 in 1H-NMR spectrum. In the same way C-4 and C-6 gave a signal at δ = 158.1 in the 13C-NMR spectrum. Furthermore, the IR spectrum showed typical absorption for the associated NH group of the pyrimidinone.
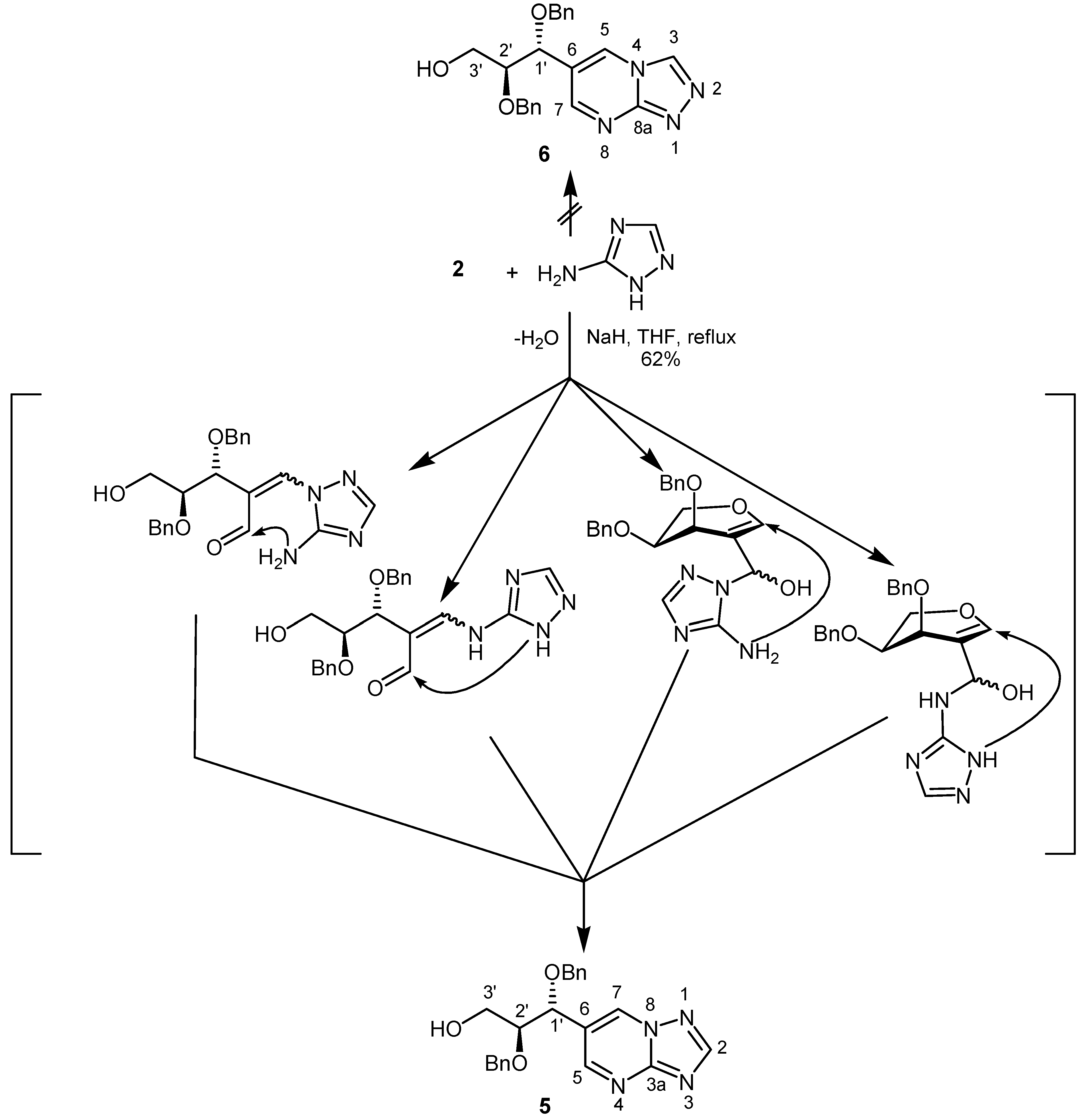
Next, 2-formyl-L-arabinal 2 was reacted with 3-amino-1,2,4-triazole in the presence of sodium hydride in tetrahydrofuran to give a fused pyrimidine. After 30 h under reflux 6-[1R,2S-1,2-bis(benzyloxy)-3-hydroxy-propyl][1,2,4]triazolo[1,5-a]pyrimidine (5) could be isolated in 62% yield (Scheme 2).
Scheme 2.
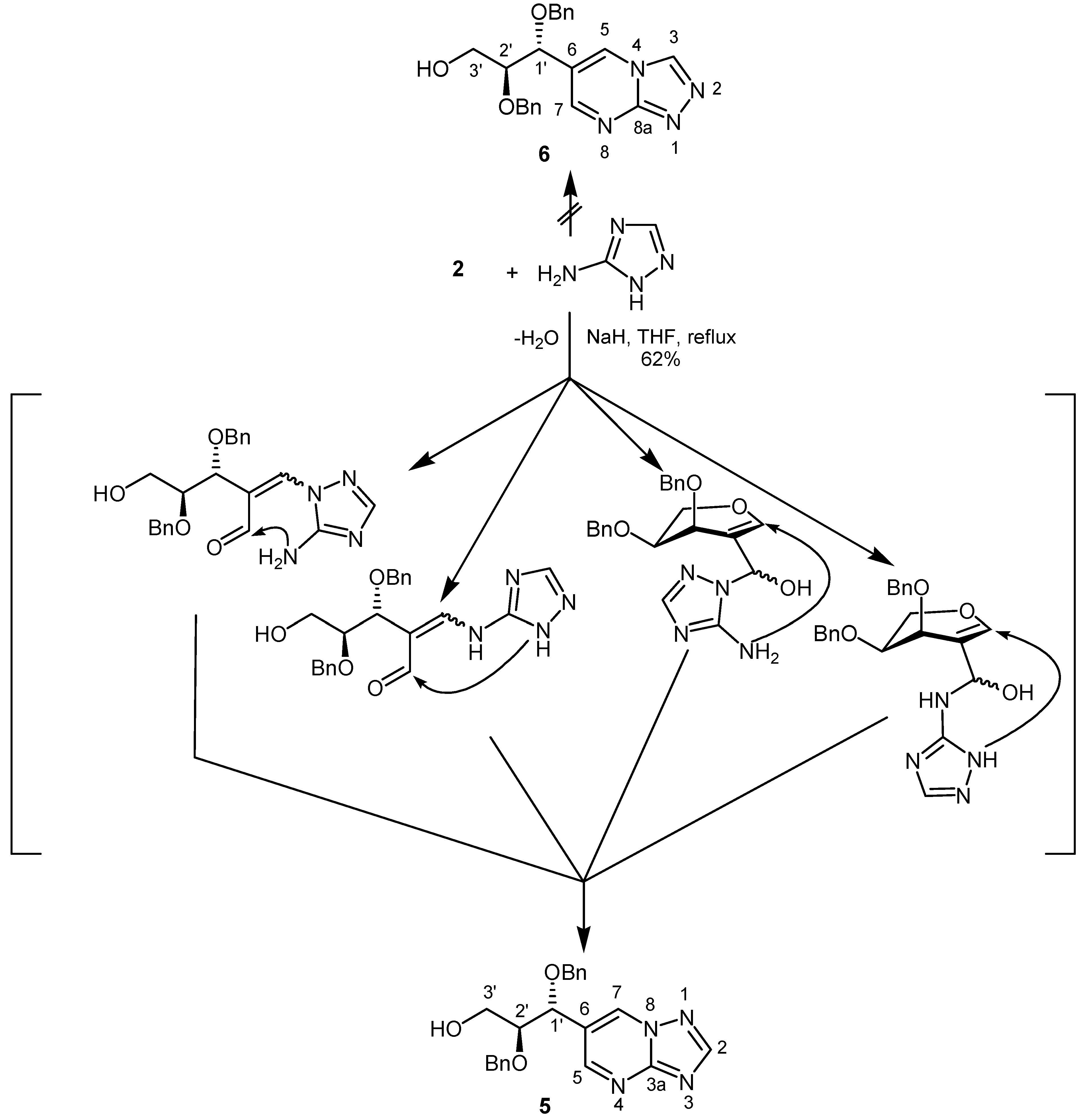
The 1H-NMR spectrum displayed two doublets for H-5 and H-7, each having a coupling constant of 2.2 Hz due to coupling over four bonds (W type). In order to distinguish between the alternative structures 5 and 6 a NOESY spectrum was recorded. The absence of cross peaks between H-3 and H-5 protons, as expected for structure 6, confirmed structure 5. On the other hand, a correlation between H‑7 and H-1´ was found.
Four conceivable pathways can be formulated for this reaction. The first nucleophilic attack of 3-amino-1,2,4-triazole may occur with the ring NH and NH2 groups, respectively, at C-1 of the 2-formyl-L-arabinal resulting in cleavage of the pyranose ring. After that recyclization is possible by reaction of the NH2 or ring NH group with the formyl function to yield [1,2,4]triazolo[1,5-a]-pyrimidine 5. On the other hand, the first nucleophilic attack of ring NH and NH2 groups, respectively, could take place at the carbonyl group of 2-formyl-L-arabinal 2 followed by ring transformation including now the NH2 and ring NH groups. Finally, all these reaction pathways result in the formation of the same [1,2,4]triazolo[1,5-a]pyrimidine 5.
Experimental
General
Solvents were distilled and if necessary, dried using standard procedures. TLC was carried out on silica gel 60 GF254 (Merck) with detection by UV light (λ = 254 nm) and/or by charring with 10% sulfuric acid in methanol. Silica gel 60 (70-230 mesh) (Merck) was used for column chromatography. Melting points were determined by using a Boetius melting point apparatus and are corrected. Specific rotations were determined with a Gyromat HP (Dr. Kernchen). IR spectra were recorded with a Nicolet 205 FT-IR spectrometer. 1H-NMR spectra (250.13 MHz and 300.13 MHz, respectively) and 13C-NMR spectra (62.9 MHz and 75.5 MHz, respectively) were recorded on Bruker instruments AC 250 and ARX 300, with CDCl3 as solvent. The calibration of spectra was carried out on the solvent signals (δ(1H) = 7.25; δ(13C) = 77.0). The 1H- and 13C-NMR signals were assigned by DEPT and two-dimensional 1H,1H Cosy and 13C,1H correlation spectra (HETCOR). The mass spectra were recorded on an AMD 402/3 spectrometer (AMD Intectra GmbH). Elemental analyses were performed on a CHNS automatic elemental analyser Flash EA 1112 (ThermoQuest).
5-[1R,2S-1,2-Bis(benzyloxy)-3-hydroxy-propyl]-1,2-dihydro-pyrimidine-2-thione (3)
To a vigorously stirred solution of 2 (100 mg, 0.3 mmol) and thiourea (46 mg, 0.60 mmol) in anhyd. THF (6 mL) was added NaH (14 mg, 0.58 mmol) at 0 oC. After 30 min the reaction mixture was allowed to warm up to room temperature and stirring was continued until no starting material could be observed in the TLC (approx. 40 h). Methanol (3 mL) was added and the mixture was stirred for further 5 min. The resulting solution was diluted with water (25 mL) and extracted with chloroform (2×25 mL). Then the combined organic layers were dried (Na2SO4), filtered and evaporated under reduced pressure. The residue obtained was purified by column chromatography (toluene/EtOAc 6:4) to give a yellow syrup. Yield: 45 mg (38%); Rf: 0.35 (toluene/EtOAc 6:4); ![Molecules 10 00837 i001]() : – 41.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3418 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.07 (br, 1Η, ΟΗ), 3.56 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.83 (d, 2H, H-3´), 4.31 (d, 1H, J = 11.5 Hz, CHHPh), 4.35 (d, 1H, J = 11.5 Hz, CHHPh), 4.42 (d, 1H, H-1´), 4.50 (d, 1H, J = 11.5 Hz, CHHPh ), 4.51 (d, 1H, J = 11.5 Hz, CHHPh), 5.00 (s, 1H, NH), 7.01–7.06 (m, 2H, Ph), 7.23–7.38 (m, 6H, Ph), 8.50 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.2 (C-3´), 71.6, 72.7 (CH2Ph), 76.7 (C-1´), 80.9 (C-2´), 125.3 (C-5), 128.0, 128.1, 128.1, 128.2, 128.5, 128.6 (Ph), 136.8, 136.9 (i-Ph), 157.0 (C-4, C-6), 170.7 (C-2); MS (CI): m/z (%): 383 (62) [M+H]+, 91 (100); Anal. Calcd. for C21H22N2O3S: C, 60.95; H, 5.80; N, 7.32; S, 8.38. Found: C, 65.86; H, 6.12; N, 7.09; S, 7.95.
: – 41.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3418 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.07 (br, 1Η, ΟΗ), 3.56 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.83 (d, 2H, H-3´), 4.31 (d, 1H, J = 11.5 Hz, CHHPh), 4.35 (d, 1H, J = 11.5 Hz, CHHPh), 4.42 (d, 1H, H-1´), 4.50 (d, 1H, J = 11.5 Hz, CHHPh ), 4.51 (d, 1H, J = 11.5 Hz, CHHPh), 5.00 (s, 1H, NH), 7.01–7.06 (m, 2H, Ph), 7.23–7.38 (m, 6H, Ph), 8.50 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.2 (C-3´), 71.6, 72.7 (CH2Ph), 76.7 (C-1´), 80.9 (C-2´), 125.3 (C-5), 128.0, 128.1, 128.1, 128.2, 128.5, 128.6 (Ph), 136.8, 136.9 (i-Ph), 157.0 (C-4, C-6), 170.7 (C-2); MS (CI): m/z (%): 383 (62) [M+H]+, 91 (100); Anal. Calcd. for C21H22N2O3S: C, 60.95; H, 5.80; N, 7.32; S, 8.38. Found: C, 65.86; H, 6.12; N, 7.09; S, 7.95.
 : – 41.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3418 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.07 (br, 1Η, ΟΗ), 3.56 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.83 (d, 2H, H-3´), 4.31 (d, 1H, J = 11.5 Hz, CHHPh), 4.35 (d, 1H, J = 11.5 Hz, CHHPh), 4.42 (d, 1H, H-1´), 4.50 (d, 1H, J = 11.5 Hz, CHHPh ), 4.51 (d, 1H, J = 11.5 Hz, CHHPh), 5.00 (s, 1H, NH), 7.01–7.06 (m, 2H, Ph), 7.23–7.38 (m, 6H, Ph), 8.50 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.2 (C-3´), 71.6, 72.7 (CH2Ph), 76.7 (C-1´), 80.9 (C-2´), 125.3 (C-5), 128.0, 128.1, 128.1, 128.2, 128.5, 128.6 (Ph), 136.8, 136.9 (i-Ph), 157.0 (C-4, C-6), 170.7 (C-2); MS (CI): m/z (%): 383 (62) [M+H]+, 91 (100); Anal. Calcd. for C21H22N2O3S: C, 60.95; H, 5.80; N, 7.32; S, 8.38. Found: C, 65.86; H, 6.12; N, 7.09; S, 7.95.
: – 41.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3418 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.07 (br, 1Η, ΟΗ), 3.56 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.83 (d, 2H, H-3´), 4.31 (d, 1H, J = 11.5 Hz, CHHPh), 4.35 (d, 1H, J = 11.5 Hz, CHHPh), 4.42 (d, 1H, H-1´), 4.50 (d, 1H, J = 11.5 Hz, CHHPh ), 4.51 (d, 1H, J = 11.5 Hz, CHHPh), 5.00 (s, 1H, NH), 7.01–7.06 (m, 2H, Ph), 7.23–7.38 (m, 6H, Ph), 8.50 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.2 (C-3´), 71.6, 72.7 (CH2Ph), 76.7 (C-1´), 80.9 (C-2´), 125.3 (C-5), 128.0, 128.1, 128.1, 128.2, 128.5, 128.6 (Ph), 136.8, 136.9 (i-Ph), 157.0 (C-4, C-6), 170.7 (C-2); MS (CI): m/z (%): 383 (62) [M+H]+, 91 (100); Anal. Calcd. for C21H22N2O3S: C, 60.95; H, 5.80; N, 7.32; S, 8.38. Found: C, 65.86; H, 6.12; N, 7.09; S, 7.95.5-[1R,2S-1,2-Bis(benzyloxy)-3-hydroxy-propyl]-1,2-dihydro-pyrimidin-2-one (4)
To a vigorously stirred solution of 2 (100 mg, 0.3 mmol) and cyanamide (29 mg, 0.69 mmol) in anhyd. THF (5 mL) was added NaH (11 mg, 0.45 mmol) at 0 oC. After 30 min the reaction mixture was allowed to warm up to room temperature and heated under reflux for 5 h. Then more cyanamide (29 mg, 0.69 mmol) was added and the mixture was refluxed for 30 h. Methanol (3 mL) was added and the mixture stirred for further 5 min. The resulting solution was diluted with water (25 mL) and extracted with chloroform (2×25 mL). The combined organic layers were dried (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by column chromatography (toluene/EtOAc 6:4) to give a colourless syrup. Yield: 34 mg (30%); Rf: 0.35 (toluene/EtOAc 6:4); ![Molecules 10 00837 i001]() : – 46.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3448 (OH, NH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.75 (br, 1H, OH), 3.58 (dt, 1H, J1´,2´ = 7.8 Hz, J2´,3´ = 4.5 Hz, H-2´), 3.80 (d, 2H, H-3´), 4.32 (d, 1H, H-1´), 4.32 (d, 1H, J = 11.6 Hz, CHHPh), 4.36 (d, 1H, J = 11.6 Hz, CHHPh), 4.49 (d, 1H, J = 11.6 Hz, CHHPh), 4.50 (d, 1H, J = 11.6 Hz, CHHPh), 5.43 (s, 1H, NH), 7.07–7.11 (m, 2H, Ph), 7.24–7.36 (m, 8H, Ph), 8.25 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.6 (C-3´), 71.0, 72.9 (CH2Ph), 77.2 (C-1´), 81.4 (C-2´), 121.8 (C-5), 127.9, 127.9, 128.0, 128.0, 128.4, 128.6 (Ph), 137.2, 137.3 (i-Ph), 158.0 (C-4, C-6), 162.8 (C=O); MS (EI): m/z (%): 366 (62) [M]+, 215 (85) ([M]–HOCH2CHOBn) +, 91 (89); Anal. Calcd. for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.58; H, 6.59; N, 7.69.
: – 46.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3448 (OH, NH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.75 (br, 1H, OH), 3.58 (dt, 1H, J1´,2´ = 7.8 Hz, J2´,3´ = 4.5 Hz, H-2´), 3.80 (d, 2H, H-3´), 4.32 (d, 1H, H-1´), 4.32 (d, 1H, J = 11.6 Hz, CHHPh), 4.36 (d, 1H, J = 11.6 Hz, CHHPh), 4.49 (d, 1H, J = 11.6 Hz, CHHPh), 4.50 (d, 1H, J = 11.6 Hz, CHHPh), 5.43 (s, 1H, NH), 7.07–7.11 (m, 2H, Ph), 7.24–7.36 (m, 8H, Ph), 8.25 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.6 (C-3´), 71.0, 72.9 (CH2Ph), 77.2 (C-1´), 81.4 (C-2´), 121.8 (C-5), 127.9, 127.9, 128.0, 128.0, 128.4, 128.6 (Ph), 137.2, 137.3 (i-Ph), 158.0 (C-4, C-6), 162.8 (C=O); MS (EI): m/z (%): 366 (62) [M]+, 215 (85) ([M]–HOCH2CHOBn) +, 91 (89); Anal. Calcd. for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.58; H, 6.59; N, 7.69.
: – 46.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3448 (OH, NH); 1H-NMR (250.13 MHz, CDCl3): δ = 2.75 (br, 1H, OH), 3.58 (dt, 1H, J1´,2´ = 7.8 Hz, J2´,3´ = 4.5 Hz, H-2´), 3.80 (d, 2H, H-3´), 4.32 (d, 1H, H-1´), 4.32 (d, 1H, J = 11.6 Hz, CHHPh), 4.36 (d, 1H, J = 11.6 Hz, CHHPh), 4.49 (d, 1H, J = 11.6 Hz, CHHPh), 4.50 (d, 1H, J = 11.6 Hz, CHHPh), 5.43 (s, 1H, NH), 7.07–7.11 (m, 2H, Ph), 7.24–7.36 (m, 8H, Ph), 8.25 (s, 2H, H-4, H-6); 13C-NMR (62.9 MHz, CDCl3): δ = 61.6 (C-3´), 71.0, 72.9 (CH2Ph), 77.2 (C-1´), 81.4 (C-2´), 121.8 (C-5), 127.9, 127.9, 128.0, 128.0, 128.4, 128.6 (Ph), 137.2, 137.3 (i-Ph), 158.0 (C-4, C-6), 162.8 (C=O); MS (EI): m/z (%): 366 (62) [M]+, 215 (85) ([M]–HOCH2CHOBn) +, 91 (89); Anal. Calcd. for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.58; H, 6.59; N, 7.69. 6-[1R,2S-1,2-Bis(benzyloxy)-3-hydroxy-propyl][1,2,4]triazolo[1,5-a]pyrimidine (5)
To a vigorously stirred solution of 2 (100 mg, 0.3 mmol) and 3-amino-1,2,4-triazole (63 mg, 0.61 mmol) in anhyd. THF (5 mL) was added NaH (8 mg, 0.33 mmol) at 0 oC. After 30 min the reaction mixture was allowed to warm up to room temperature and then heated under reflux for 30 h. Methanol (3 mL) was added and the mixture was stirred for further 5 min. The resulting solution was diluted with water (25 mL) and extracted with chloroform (2×25 mL). Then the combined organic layers were dried (Na2SO4), filtered and evaporated under reduced pressure. The residue obtained was purified by column chromatography (toluene/EtOAc 6:4) to give a colourless syrup. Yield: 75 mg (62%); Rf: 0.50 (toluene/EtOAc 6:4); ![Molecules 10 00837 i001]() : – 30.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3373 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 3.21 (br, 1H, OH), 3.60 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.88 (m, center of AB part of ABX, J3´a,3´b = 11.8 Hz, 2H, H-3´a, H-3´b), 4.28 (d, 1H, J = 11.9 Hz, CHHPh), 4.43 (d, 1H, J = 11.3 Hz, CHHPh), 4.53 (d, 1H, J = 11.9 Hz, CHHPh), 4.53 (d, 1H, J = 11.3 Hz, CHHPh), 4.63 (d, 1H, H-1´), 6.90–6.93 (m, 2H, Ph), 7.00–7.09 (m, 3H, Ph), 7.22–7.33 (m, 5H, Ph), 8.49 (s, 1H, H-2), 8.65 (d, 1H, H-7), 8.74 (d, 1H, J5,7 = 2.2 Hz, H-5); 13C-NMR (62.9 MHz, CDCl3): δ = 60.6 (C-3´), 72.1, 72.5 (CH2Ph), 75.9 (C-1´), 80.5 (C-2´), 123.3 (C-6), 127.9, 128.0, 128.2, 128.2, 128.3, 128.7 (Ph), 134.5 (C-7), 1.36.4, 136.5 (i-Ph), 154.8 (C-3a), 155.5 (C-5), 156.1 (C-2); MS (CI): m/z (%): 391 (90) [M+H]+, 91 (100); Anal. Calcd. for C22H22N4O3: C, 67.69; H, 5.64; N, 14.35. Found: C, 67.35; H, 5.71; N, 14.45.
: – 30.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3373 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 3.21 (br, 1H, OH), 3.60 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.88 (m, center of AB part of ABX, J3´a,3´b = 11.8 Hz, 2H, H-3´a, H-3´b), 4.28 (d, 1H, J = 11.9 Hz, CHHPh), 4.43 (d, 1H, J = 11.3 Hz, CHHPh), 4.53 (d, 1H, J = 11.9 Hz, CHHPh), 4.53 (d, 1H, J = 11.3 Hz, CHHPh), 4.63 (d, 1H, H-1´), 6.90–6.93 (m, 2H, Ph), 7.00–7.09 (m, 3H, Ph), 7.22–7.33 (m, 5H, Ph), 8.49 (s, 1H, H-2), 8.65 (d, 1H, H-7), 8.74 (d, 1H, J5,7 = 2.2 Hz, H-5); 13C-NMR (62.9 MHz, CDCl3): δ = 60.6 (C-3´), 72.1, 72.5 (CH2Ph), 75.9 (C-1´), 80.5 (C-2´), 123.3 (C-6), 127.9, 128.0, 128.2, 128.2, 128.3, 128.7 (Ph), 134.5 (C-7), 1.36.4, 136.5 (i-Ph), 154.8 (C-3a), 155.5 (C-5), 156.1 (C-2); MS (CI): m/z (%): 391 (90) [M+H]+, 91 (100); Anal. Calcd. for C22H22N4O3: C, 67.69; H, 5.64; N, 14.35. Found: C, 67.35; H, 5.71; N, 14.45.
: – 30.1 (c = 1.0, CHCl3); IR (capillary, cm–1): 3373 (OH); 1H-NMR (250.13 MHz, CDCl3): δ = 3.21 (br, 1H, OH), 3.60 (dt, 1H, J1´,2´ = 8.0 Hz, J2´,3´ = 4.0 Hz, H-2´), 3.88 (m, center of AB part of ABX, J3´a,3´b = 11.8 Hz, 2H, H-3´a, H-3´b), 4.28 (d, 1H, J = 11.9 Hz, CHHPh), 4.43 (d, 1H, J = 11.3 Hz, CHHPh), 4.53 (d, 1H, J = 11.9 Hz, CHHPh), 4.53 (d, 1H, J = 11.3 Hz, CHHPh), 4.63 (d, 1H, H-1´), 6.90–6.93 (m, 2H, Ph), 7.00–7.09 (m, 3H, Ph), 7.22–7.33 (m, 5H, Ph), 8.49 (s, 1H, H-2), 8.65 (d, 1H, H-7), 8.74 (d, 1H, J5,7 = 2.2 Hz, H-5); 13C-NMR (62.9 MHz, CDCl3): δ = 60.6 (C-3´), 72.1, 72.5 (CH2Ph), 75.9 (C-1´), 80.5 (C-2´), 123.3 (C-6), 127.9, 128.0, 128.2, 128.2, 128.3, 128.7 (Ph), 134.5 (C-7), 1.36.4, 136.5 (i-Ph), 154.8 (C-3a), 155.5 (C-5), 156.1 (C-2); MS (CI): m/z (%): 391 (90) [M+H]+, 91 (100); Anal. Calcd. for C22H22N4O3: C, 67.69; H, 5.64; N, 14.35. Found: C, 67.35; H, 5.71; N, 14.45. Acknowledgments
We thank the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie for financial support.
References
- Borrmann, D. Houben-Weyl, Methoden der Organischen Chemie; Müller, E., Ed.; Thieme: Stuttgart, 1969; Vol. VII/4. [Google Scholar]
- Schaumann, E. Houben-Weyl, Methoden der Organischen Chemie; Klamann, D., Ed.; Thieme: Stuttgart, New York, 1985; Vol. E 11. [Google Scholar]
- Tominaga, Y. J. Heterocycl. Chem. 1989, 26, 1167–1204.
- Peseke, K.; Feist, H.; Quincoces, J. Targ. Heterocycl. Syst. 2001, 5, 299–325.
- Ramesh, N. G.; Balasubramanian, K. K. Tetrahedron Lett. 1991, 32, 3875–3878.
- Rudloff, I.; Peseke, K.; Reinke, H. J. Prakt. Chem. 1998, 340, 334–340.
- Ramesh, N. G.; Balasubramanian, K. K. Eur. J. Org. Chem. 2003, 4477–4487.
- Montero, A.; Feist, H.; Michalik, M.; Quincoces, J.; Peseke, K. Synthesis 2002, 664–669.
- Montero, A.; Feist, H.; Michalik, M.; Quincoces, J.; Peseke, K. J. Carbohydr. Chem. 2002, 21, 305–312. [CrossRef]
- Rudloff, I.; Bari, A.; Feist, H.; Michalik, M.; Reinke, H.; Peseke, K. Z. Naturforsch. 2004, 59b, 398–405.
- Montero, A.; Michalik, M.; Feist, H.; Reinke, H.; Rudloff, I.; Peseke, K. J. Carbohydr. Chem. 2004, 23, 317–327. [CrossRef]
- Bari, A.; Feist, H.; Michalik, D.; Michalik, M.; Peseke, K. Synthesis 2004, 2863–2868.
- Sample Availability: Not available.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Bari, A.; Feist, H.; Michalik, M.; Peseke, K. Pyrimidine Acyclo-C-Nucleosides by Ring Transformations of 2-Formyl-L-arabinal. Molecules 2005, 10, 837-842. https://doi.org/10.3390/10080837
AMA Style
Bari A, Feist H, Michalik M, Peseke K. Pyrimidine Acyclo-C-Nucleosides by Ring Transformations of 2-Formyl-L-arabinal. Molecules. 2005; 10(8):837-842. https://doi.org/10.3390/10080837
Chicago/Turabian StyleBari, Ahmed, Holger Feist, Manfred Michalik, and Klaus Peseke. 2005. "Pyrimidine Acyclo-C-Nucleosides by Ring Transformations of 2-Formyl-L-arabinal" Molecules 10, no. 8: 837-842. https://doi.org/10.3390/10080837