Reactivity and Biological Activity of the Marine Sesquiterpene Hydroquinone Avarol and Related Compounds from Sponges of the Order Dictyoceratida

Abstract
:Contents
- Introduction
- Quinones with a rearranged drimane skeleton from sponges of the order Dictyoceratida
- Structure
- Biosynthesis
- Reactivity
- Biological effects
- Marine pharmacology: perspectives
1. Introduction
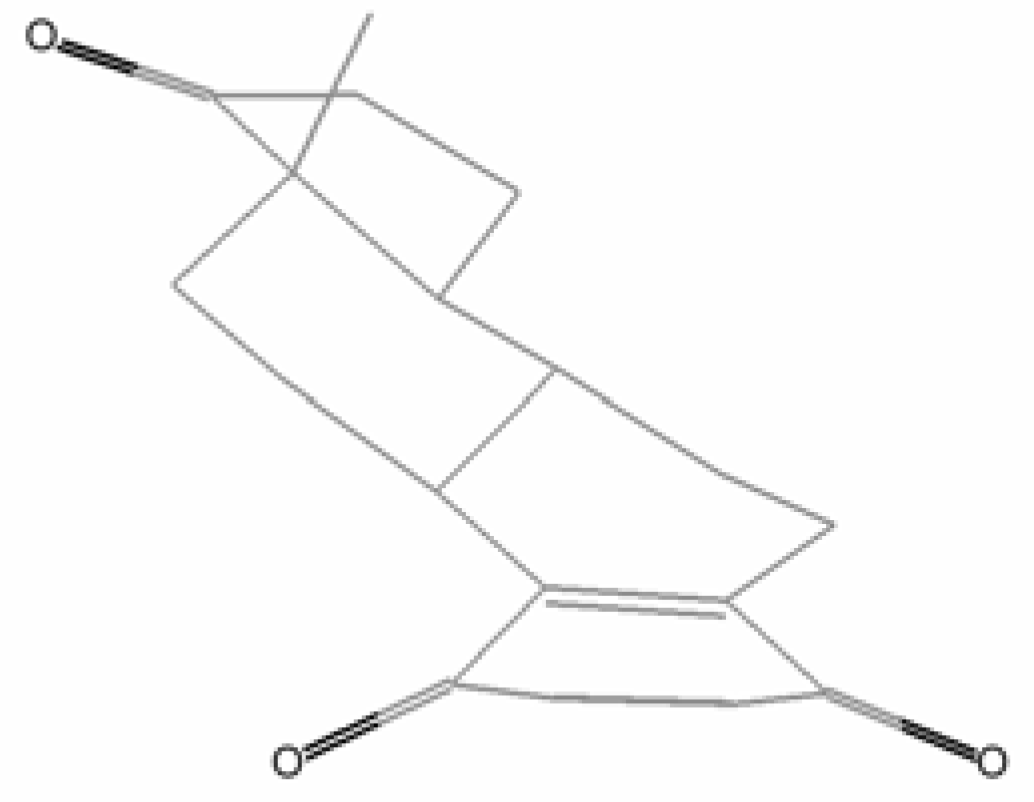
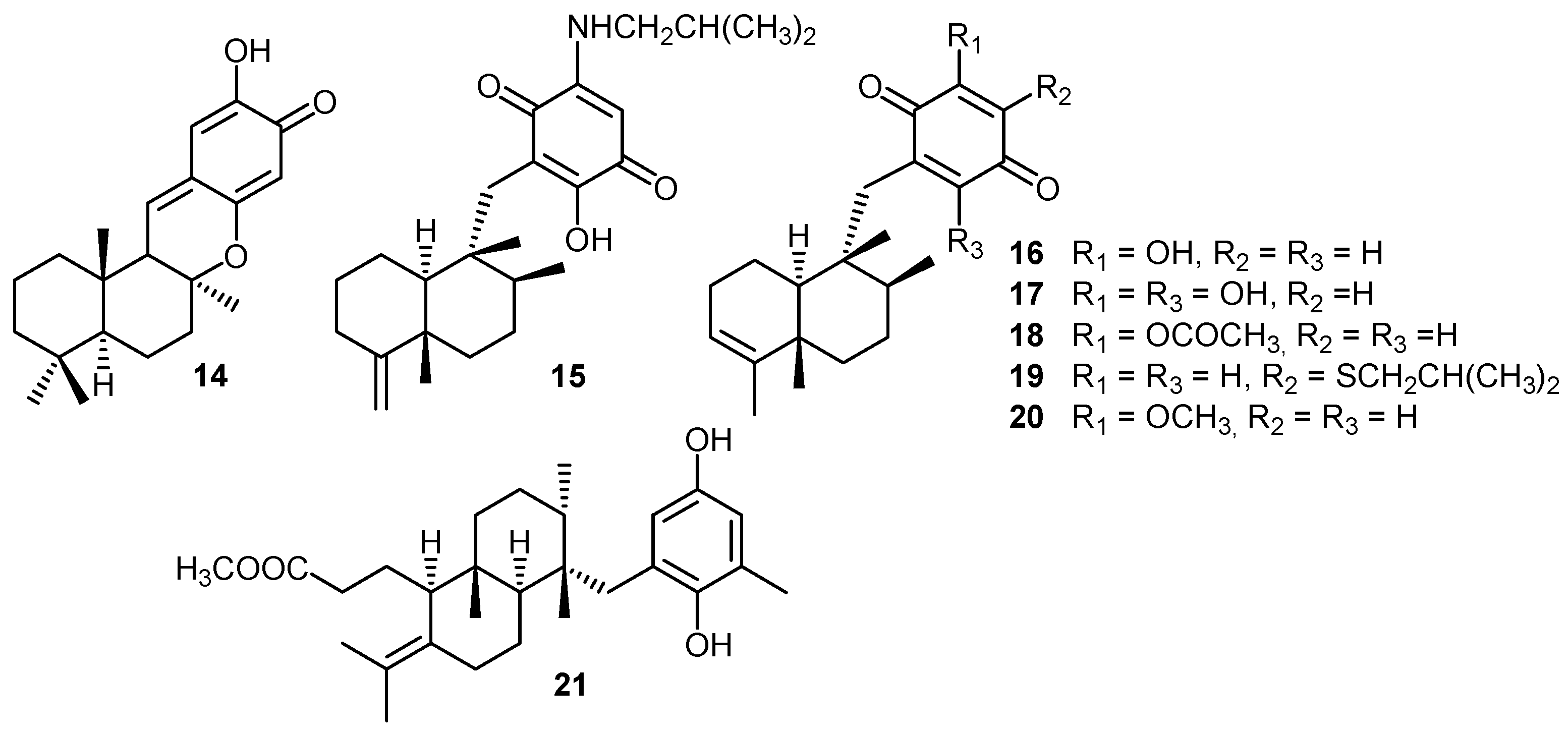
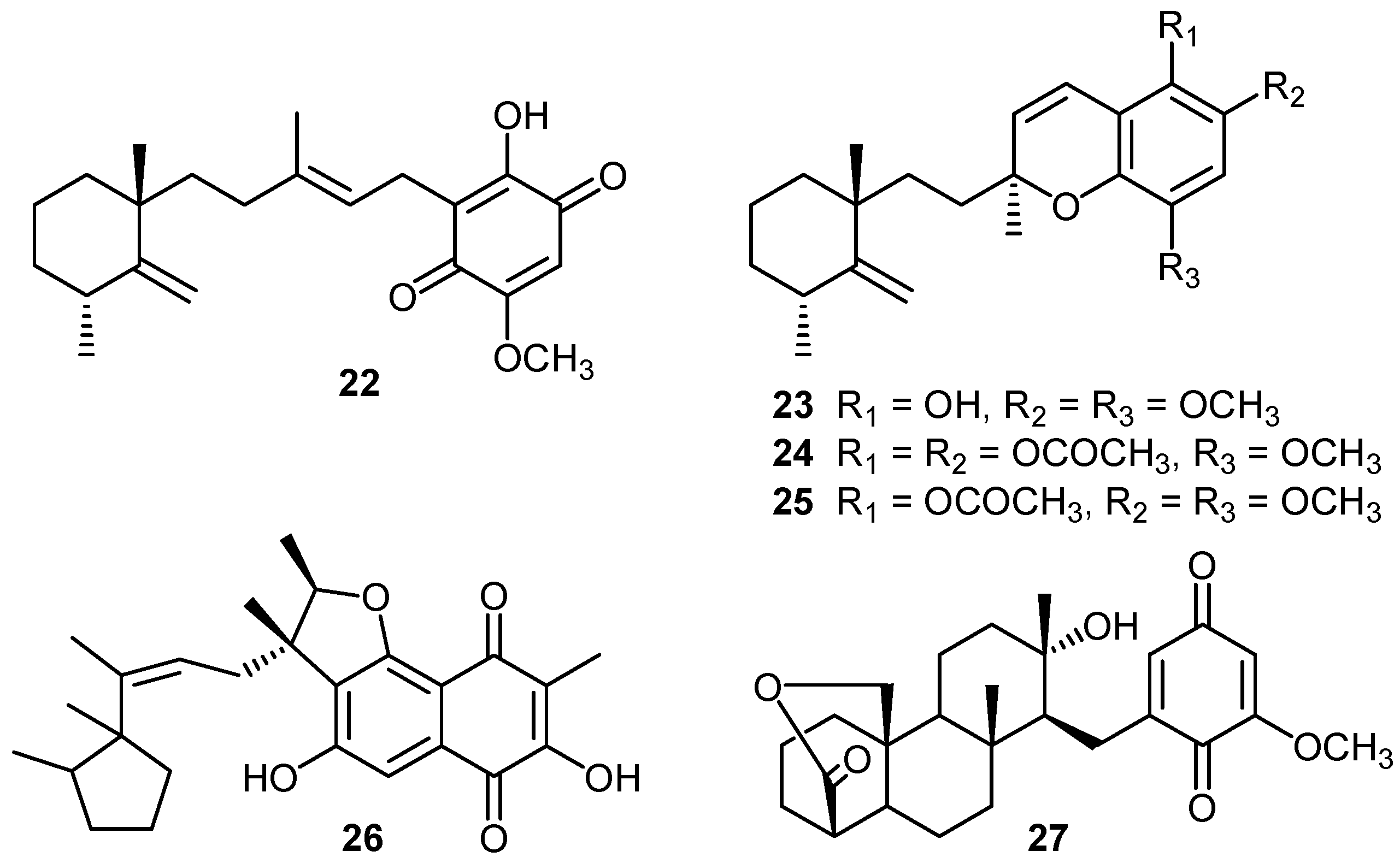
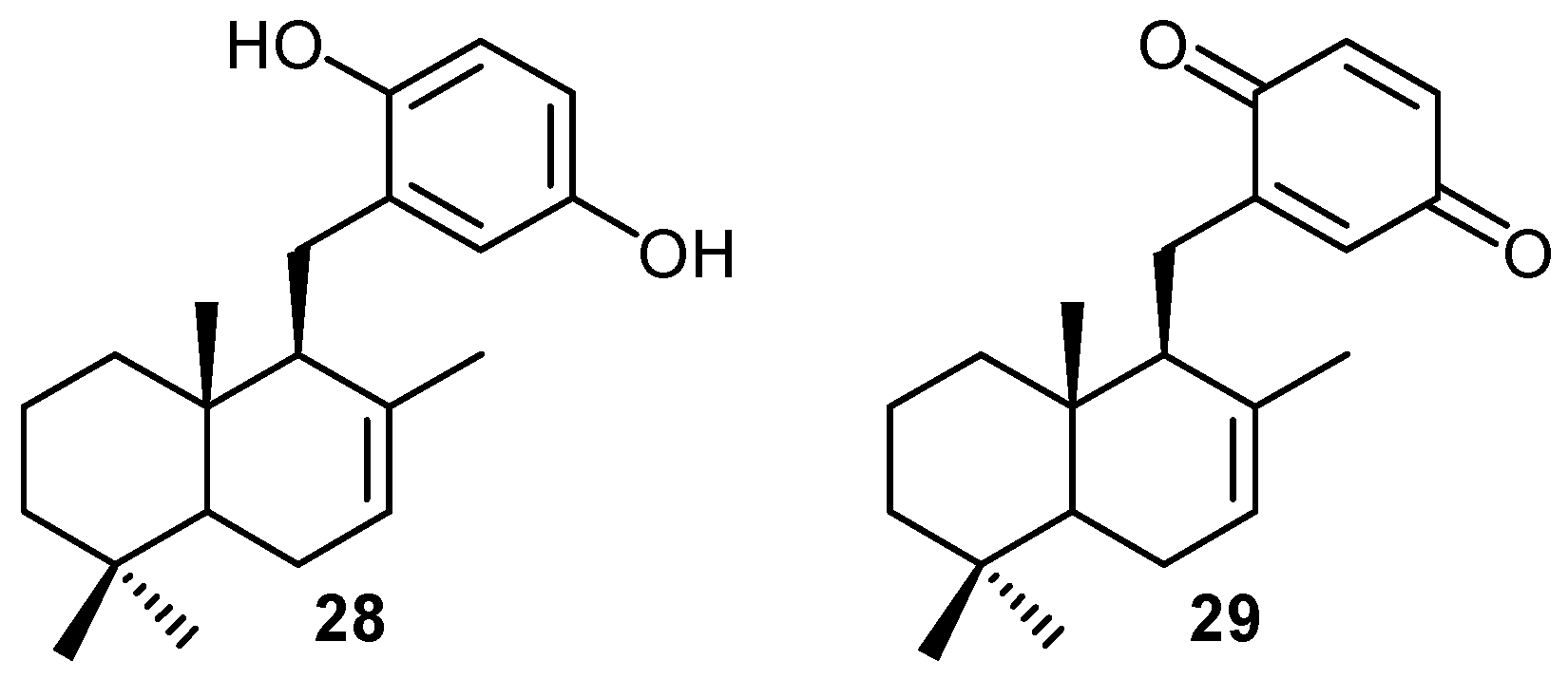
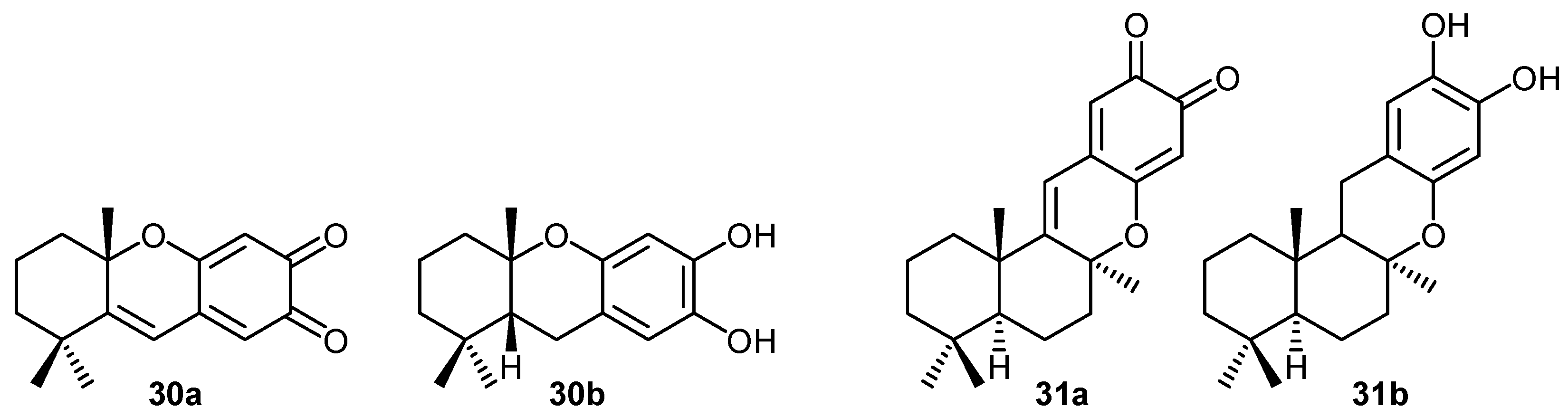
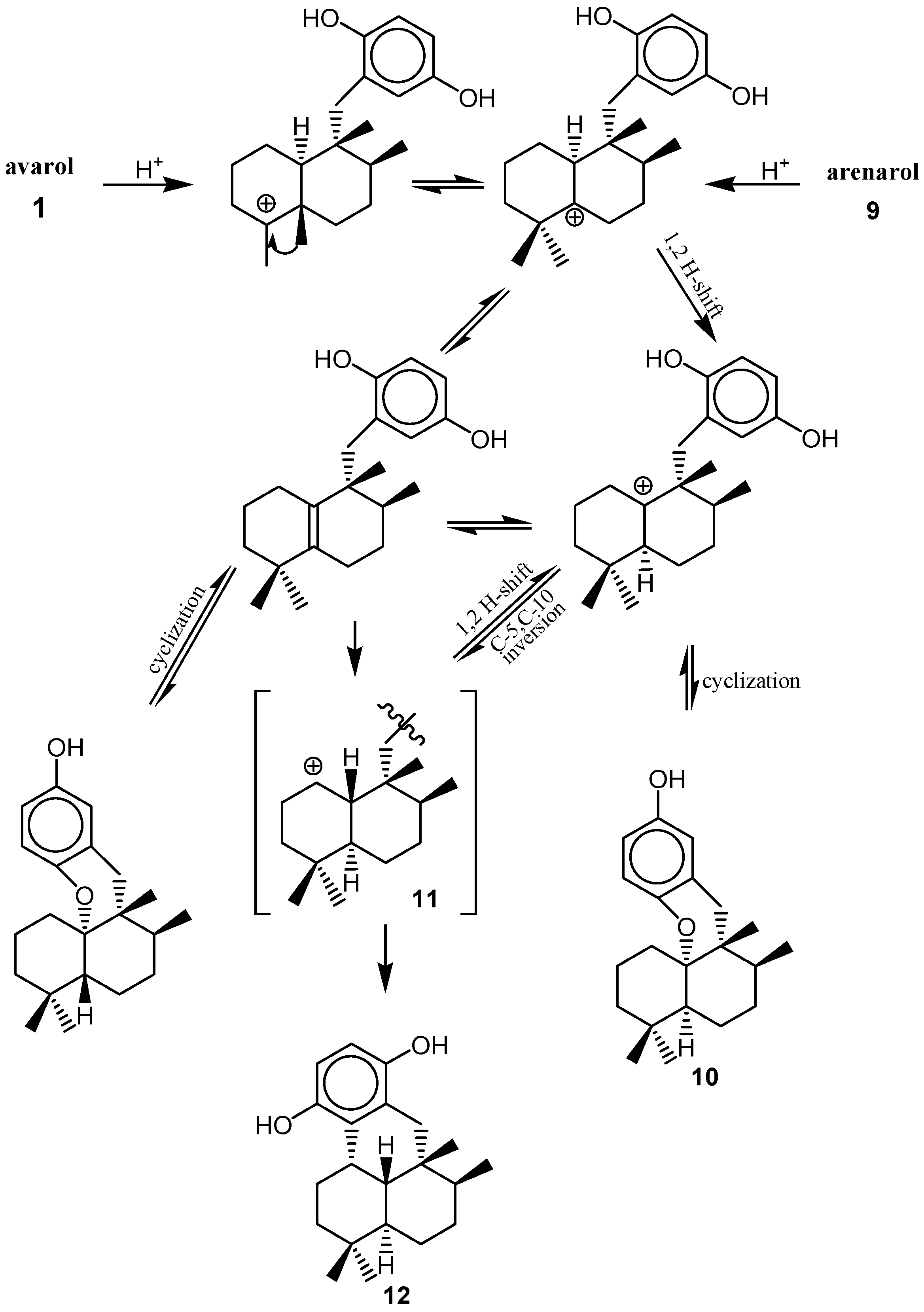
2. Quinones with a rearranged drimane skeleton from sponges of the order Dictyoceratida

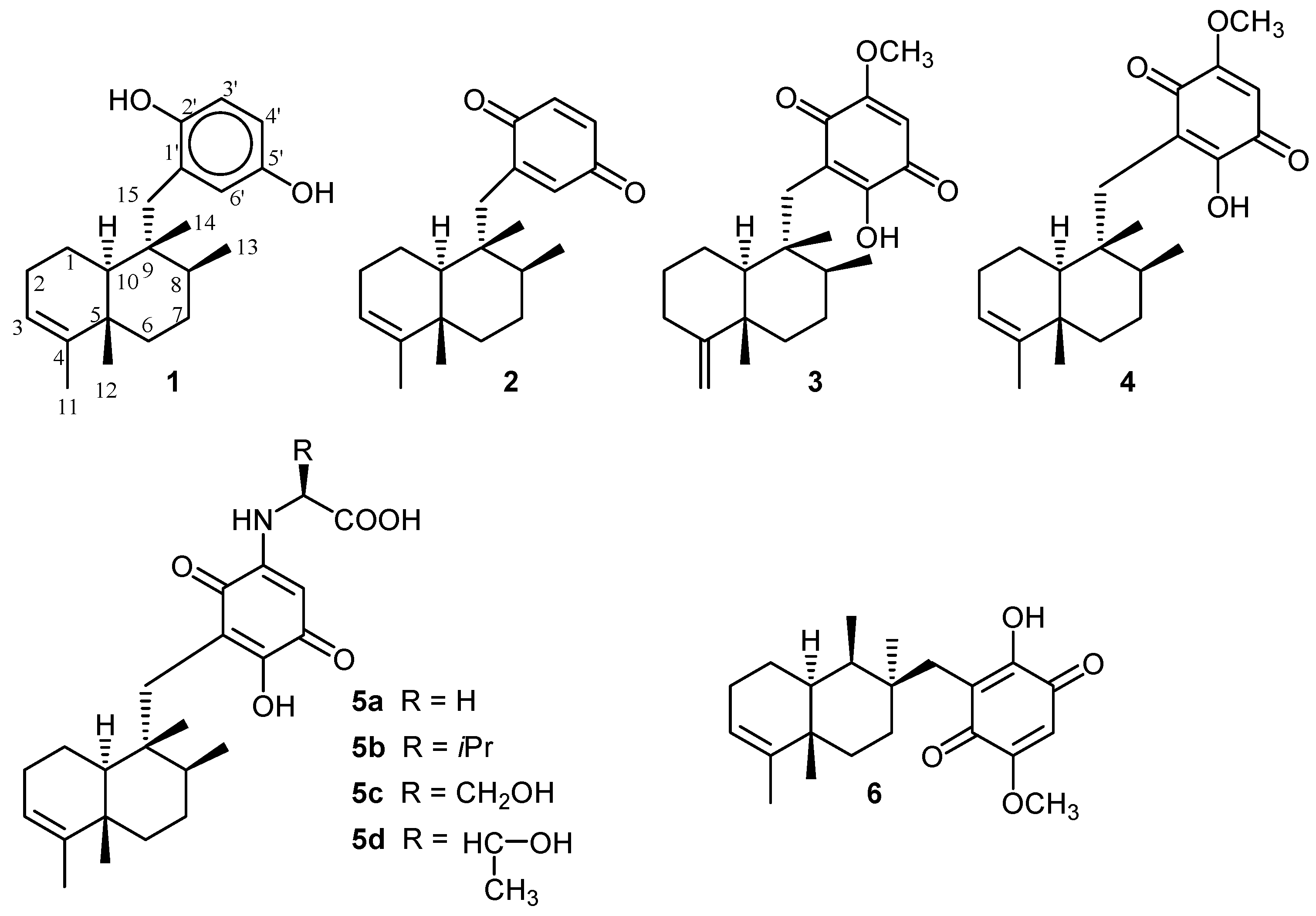
3. Structure



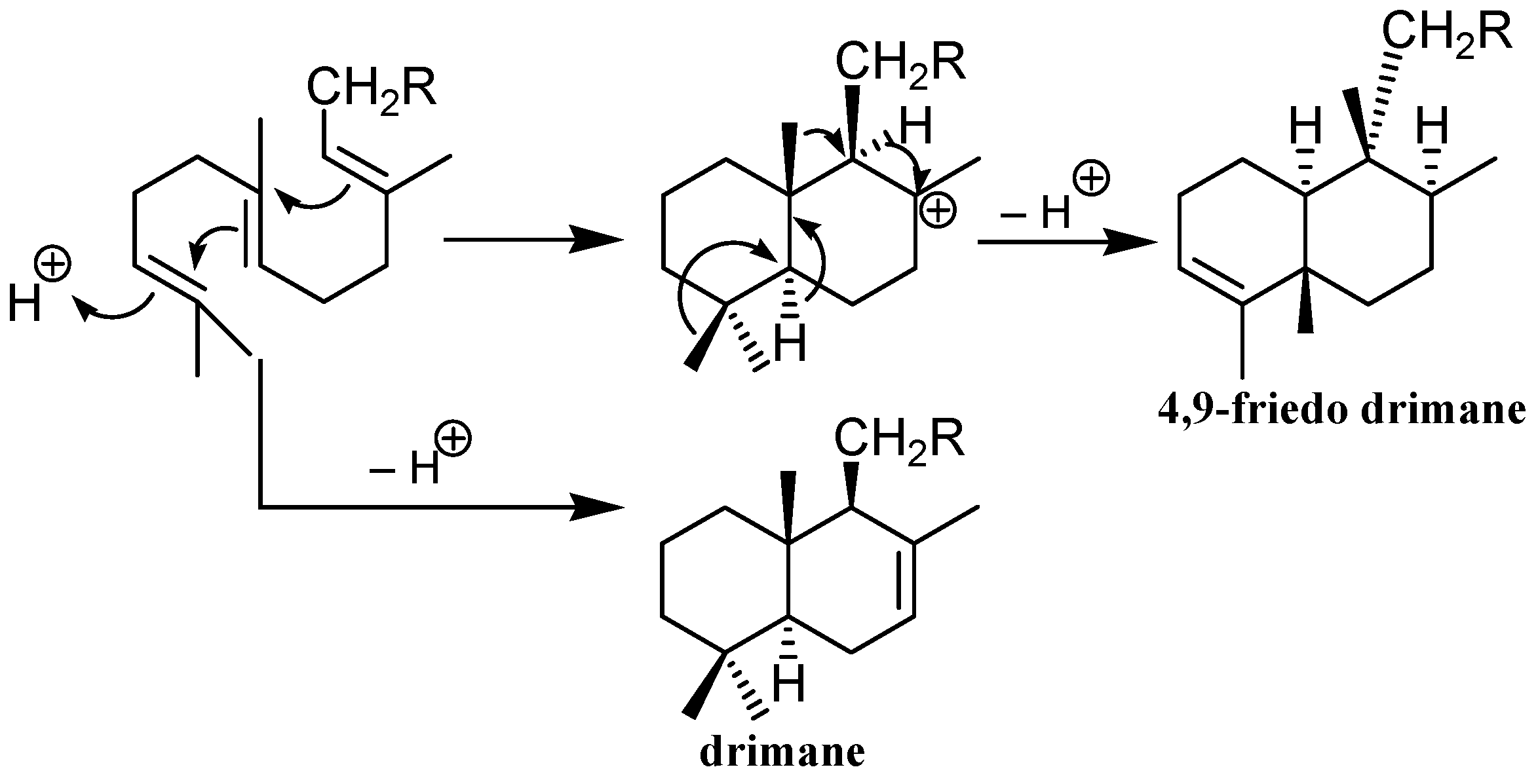
4. Biosynthesis

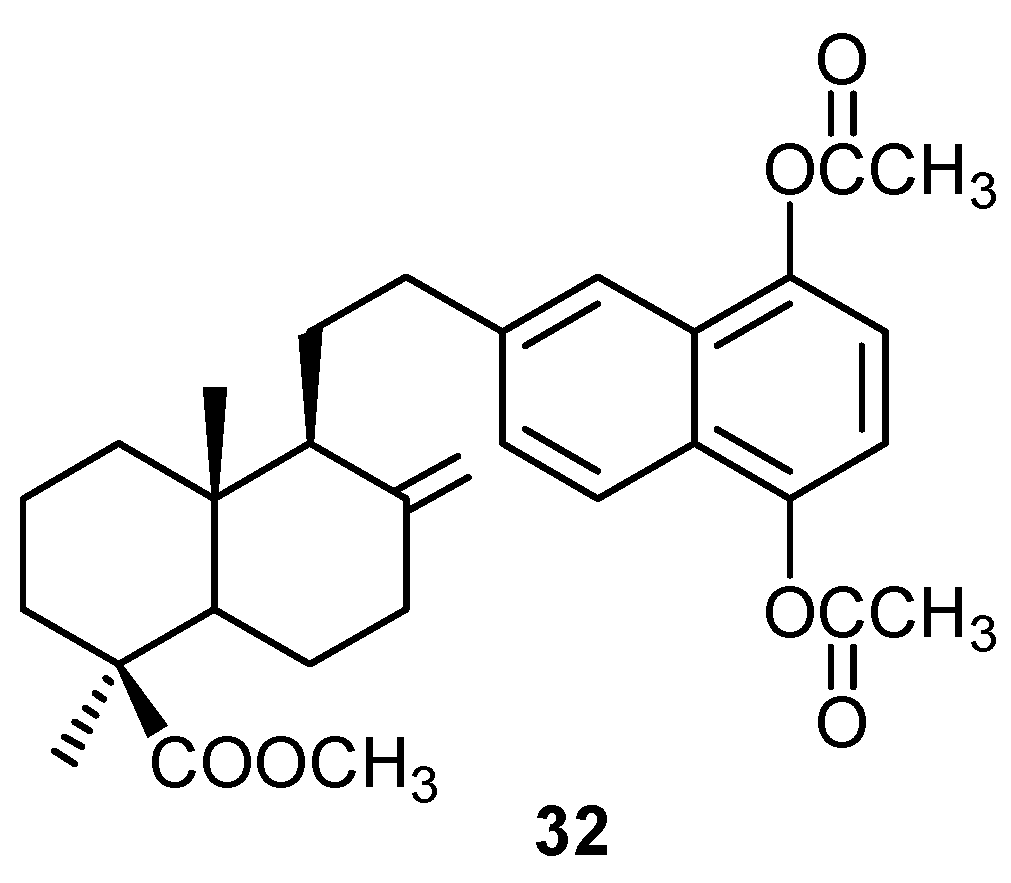
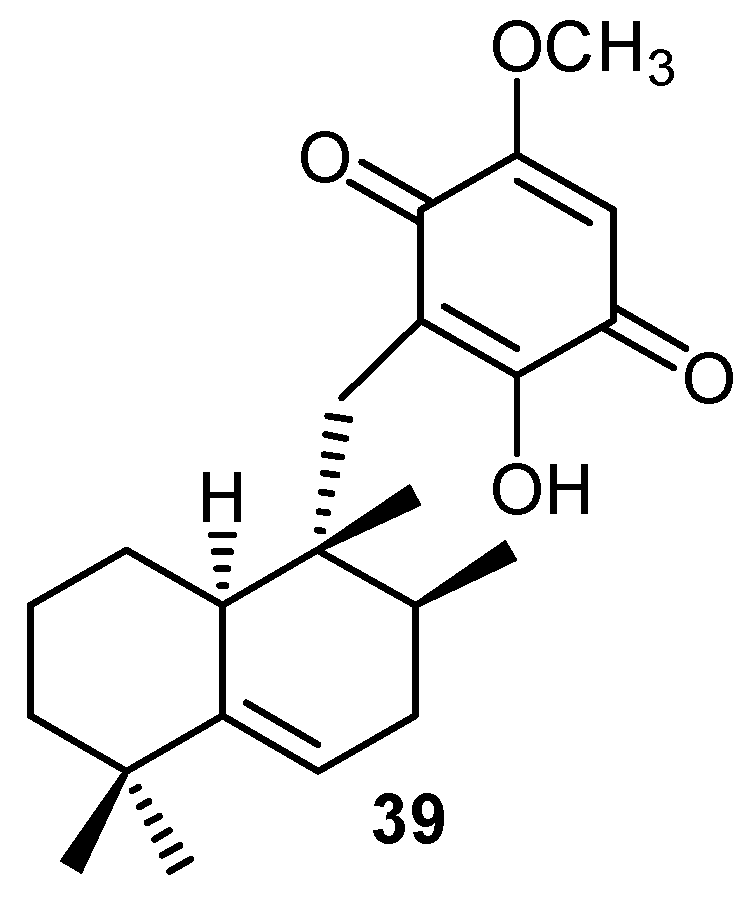
5. Reactivity
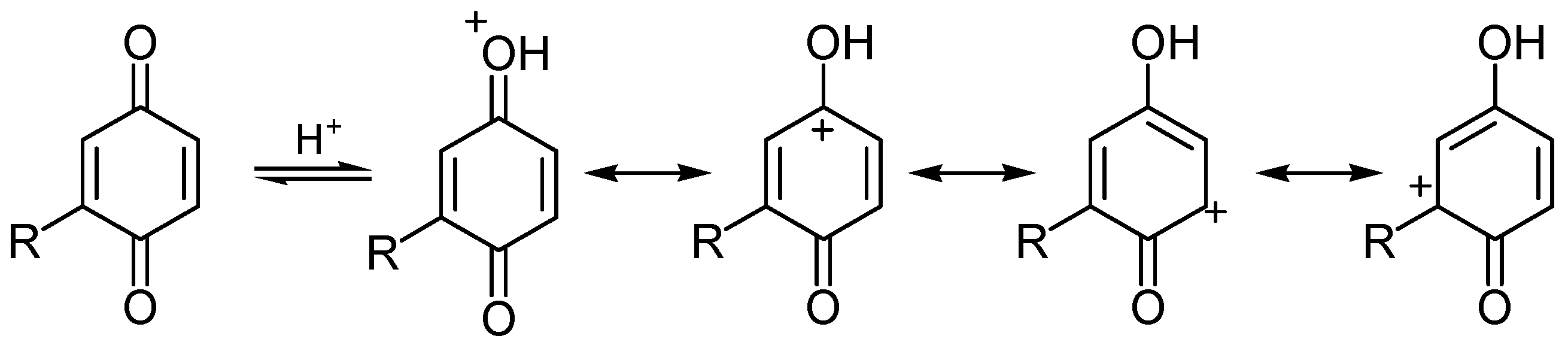
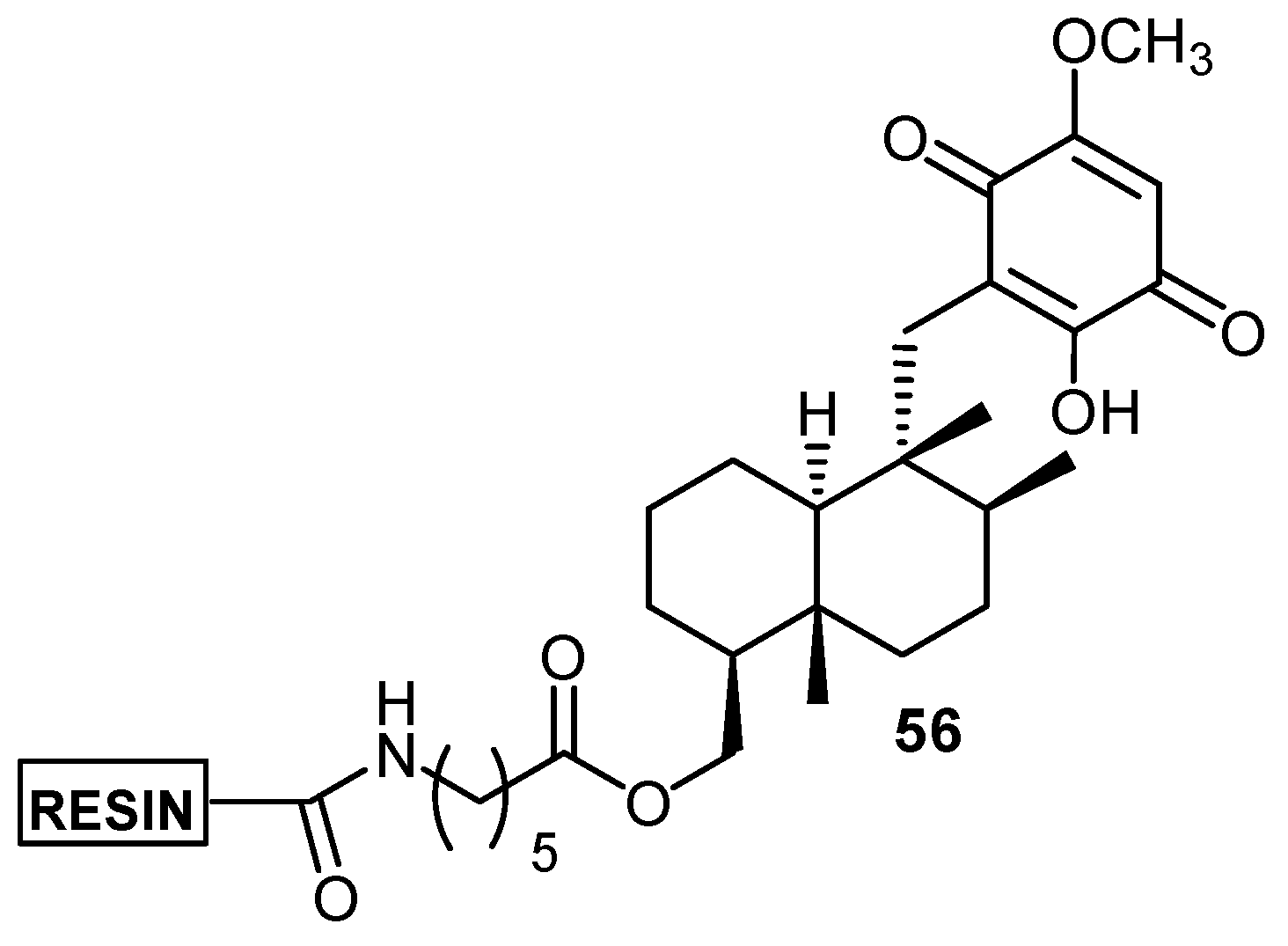
i) Redox reactivity

ii) Addition reactions
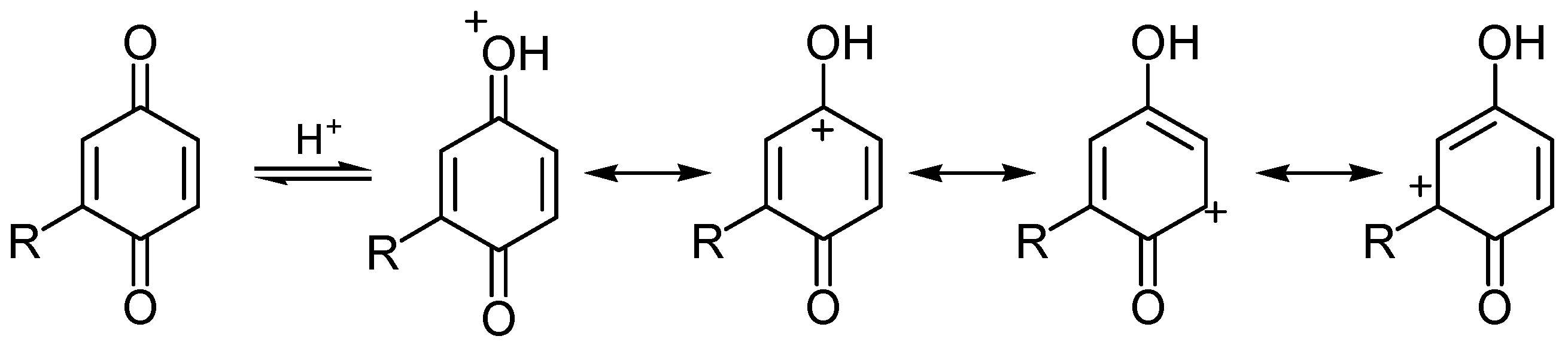


6. Biological effects
i) Antitumor activity





ii) Mechanisms of cytotoxic activity
General mechanisms
- 1)
- redox cycling of quinones, resulting in generation of reactive oxygen species which can damage biomolecules, and inhibition of the mitochondrial electron transport and/or of oxidative phosphorylation;
- 2)
- electrophilic arylation of critical cellular nucleophiles, either by quinones or quinone methides formed after bioreductive activation of quinones [65].
DNA damage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
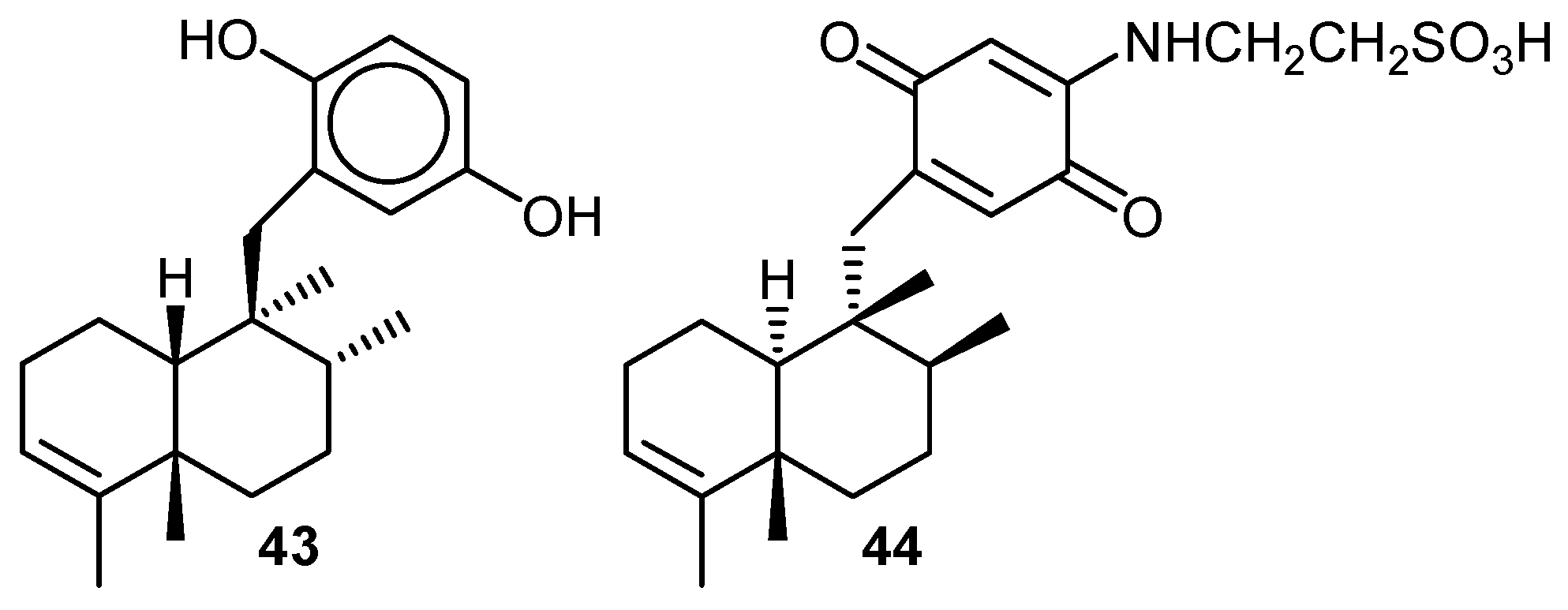{kind=link}
{kind=link}
{kind=link}
{kind=link}
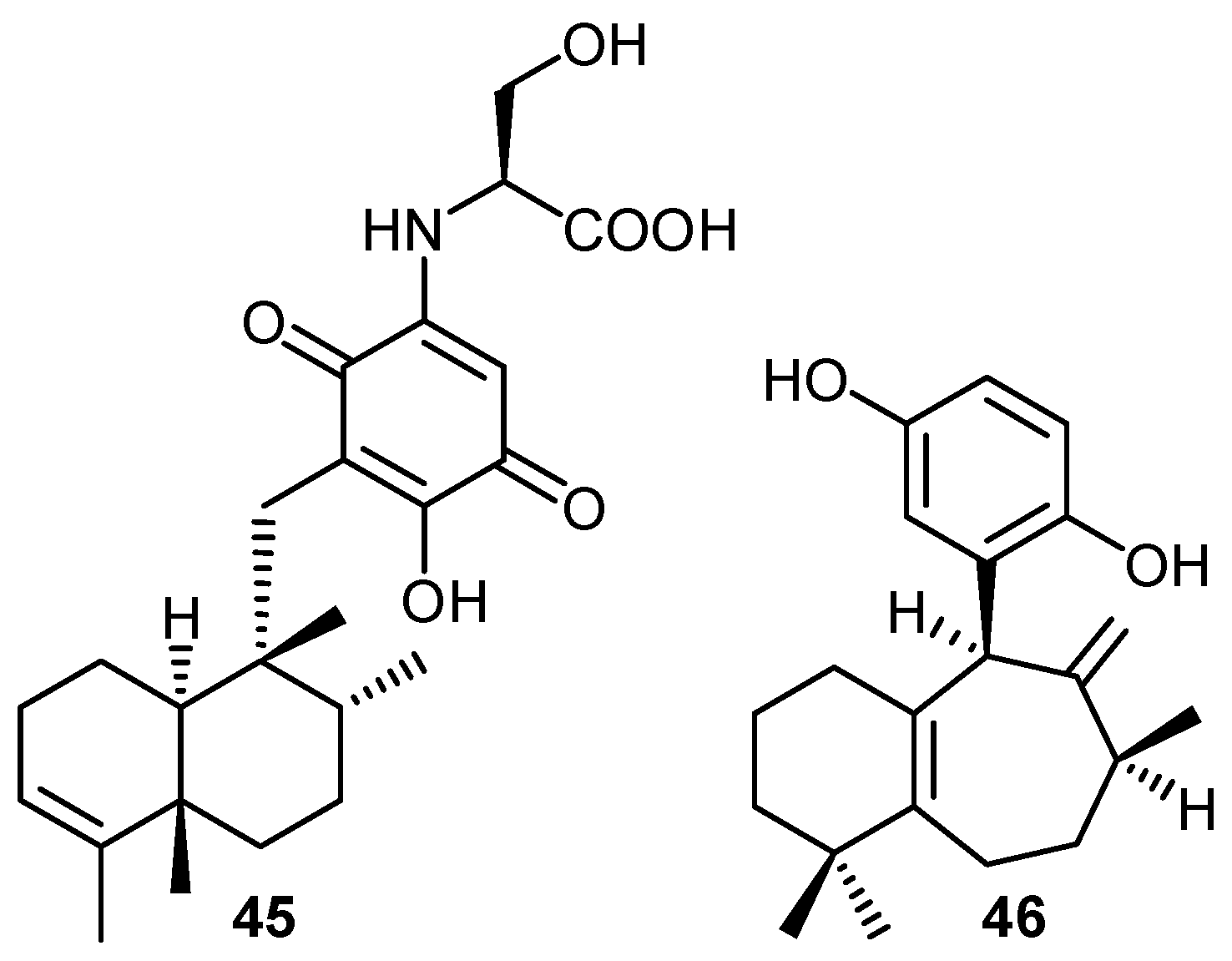
{kind=link}
{kind=link}
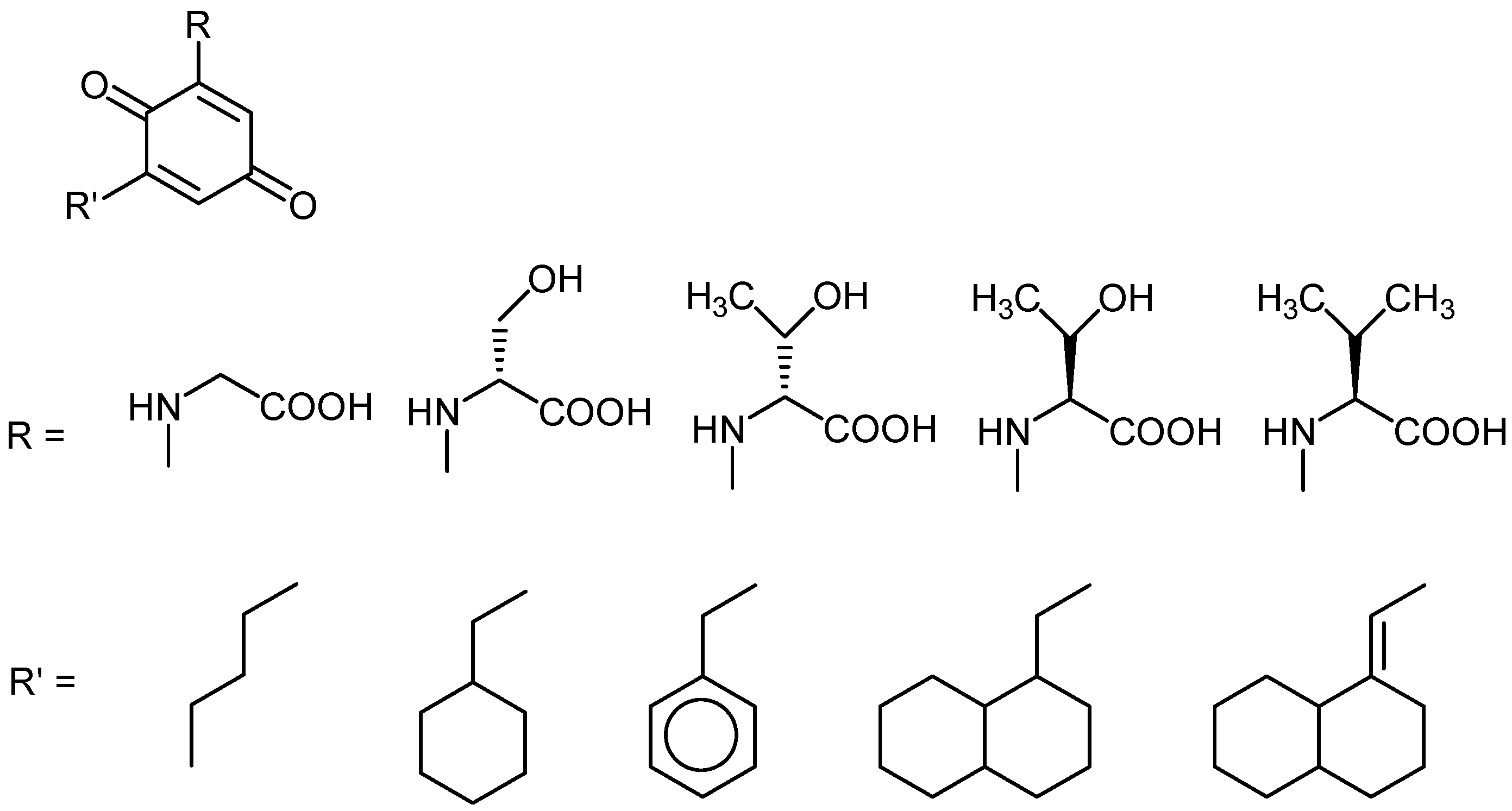{kind=link}
{kind=link}
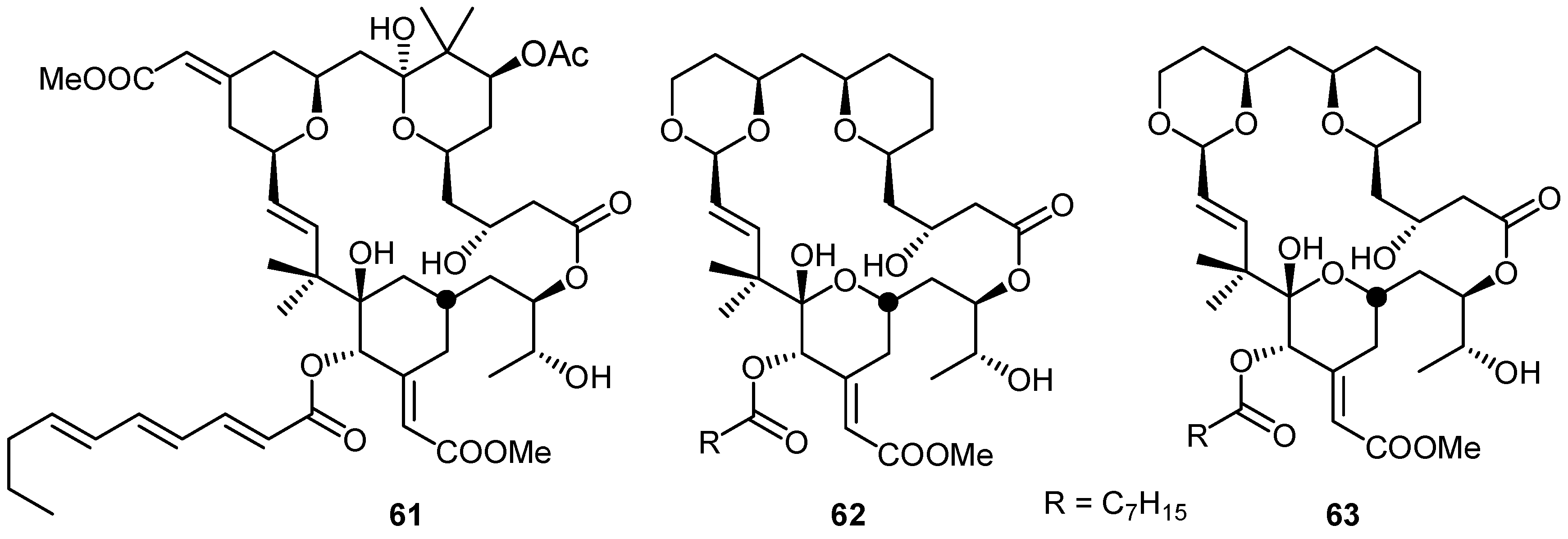{kind=link}
{kind=link}
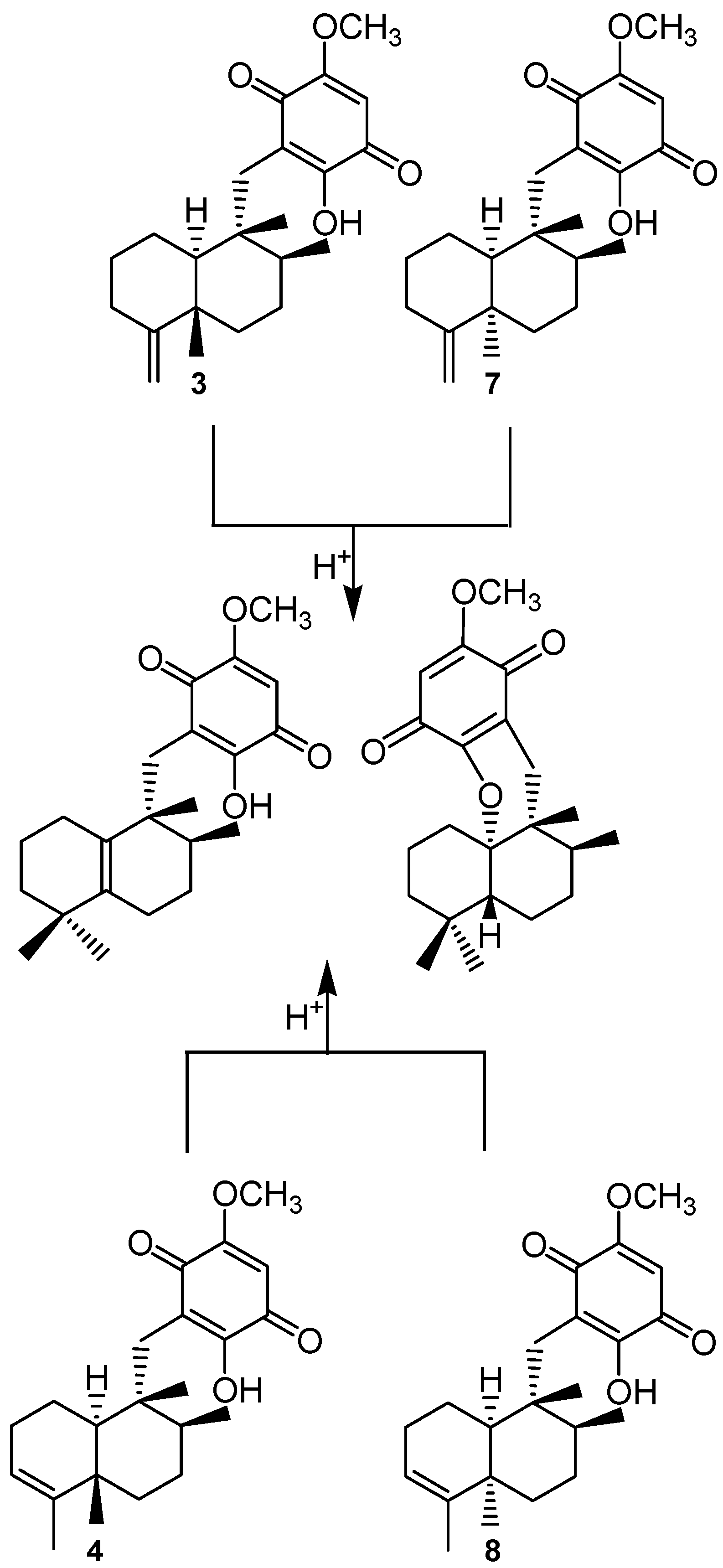{kind=link}
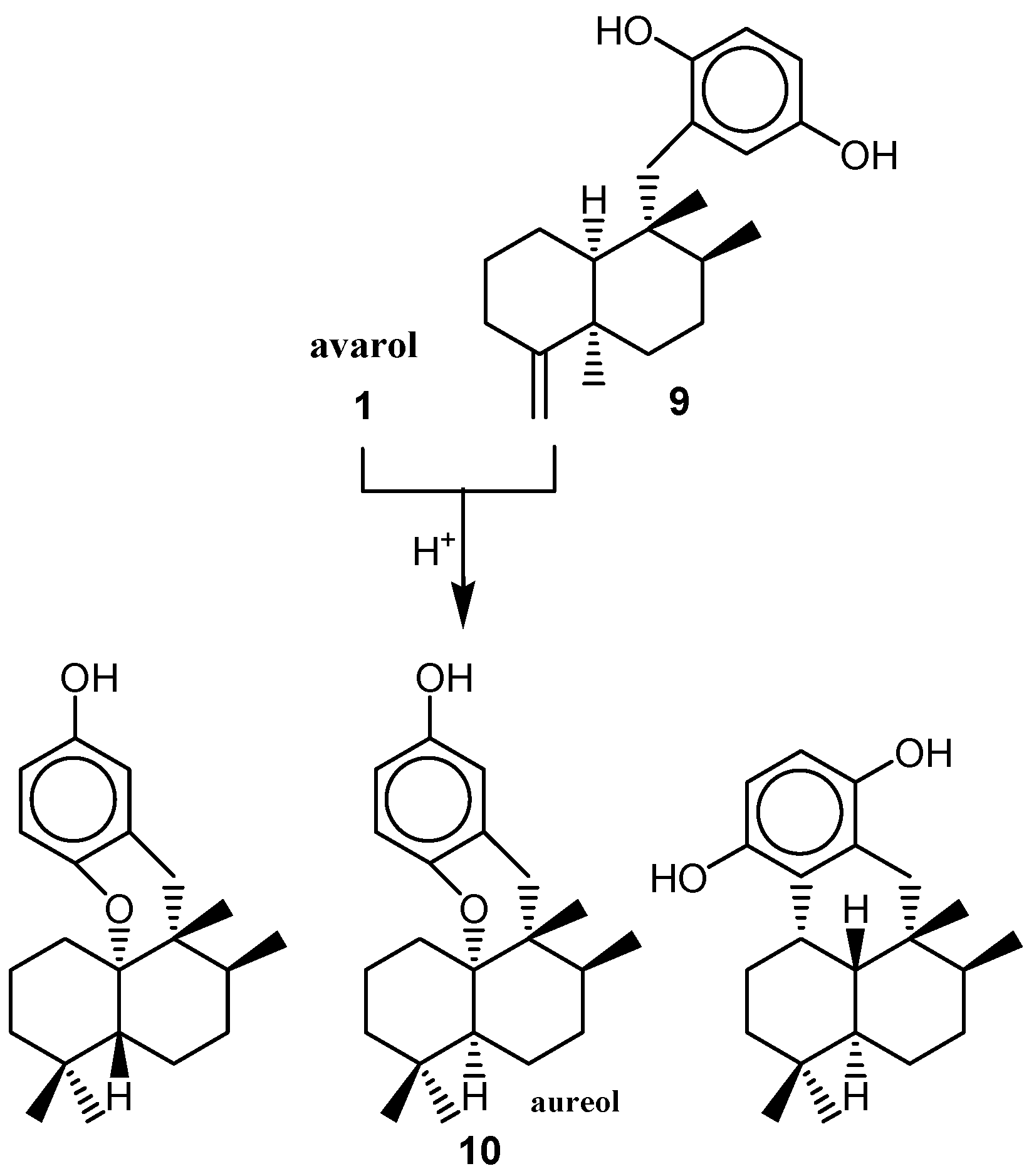{kind=link}
{kind=link}
{kind=link}
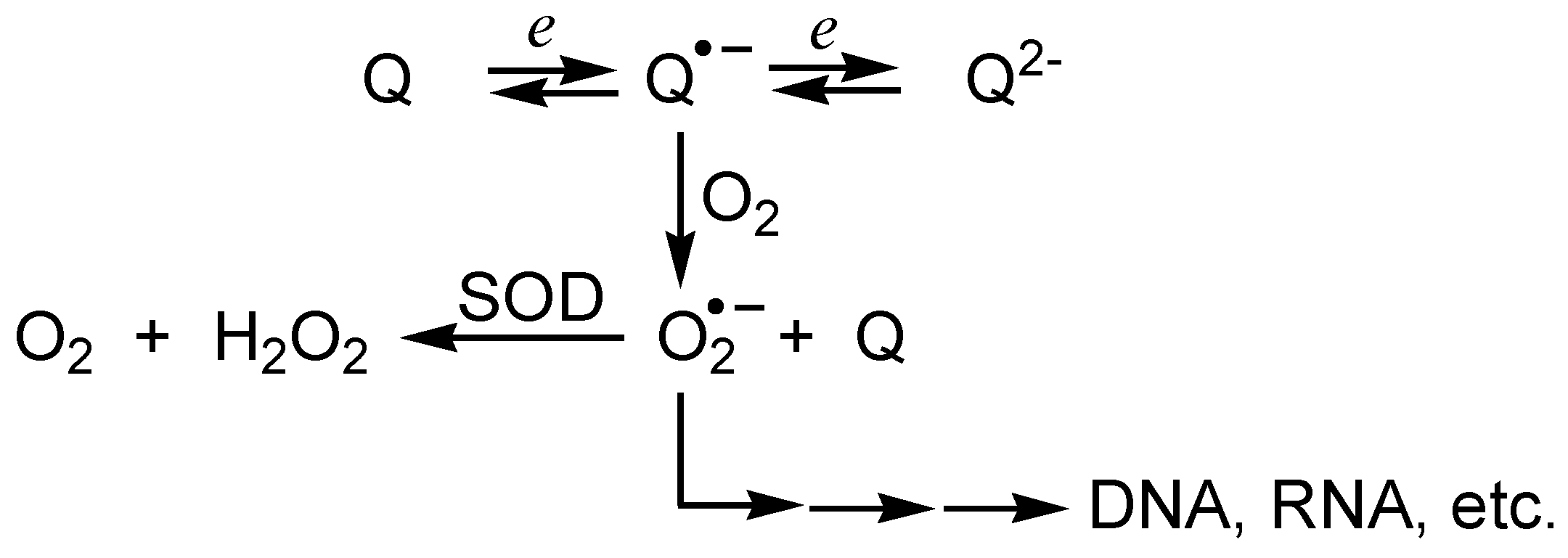{kind=link}
{kind=link}
{kind=link}
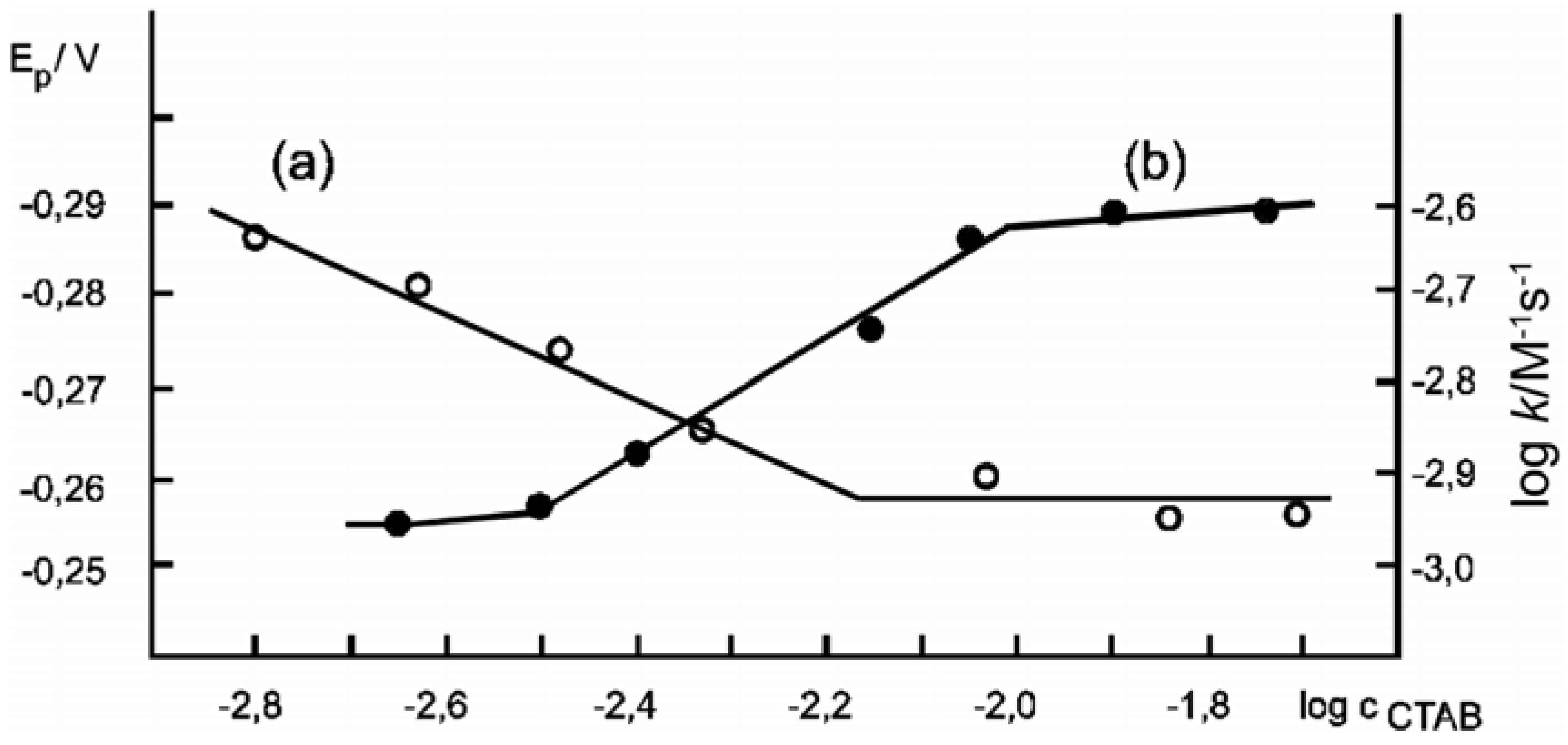
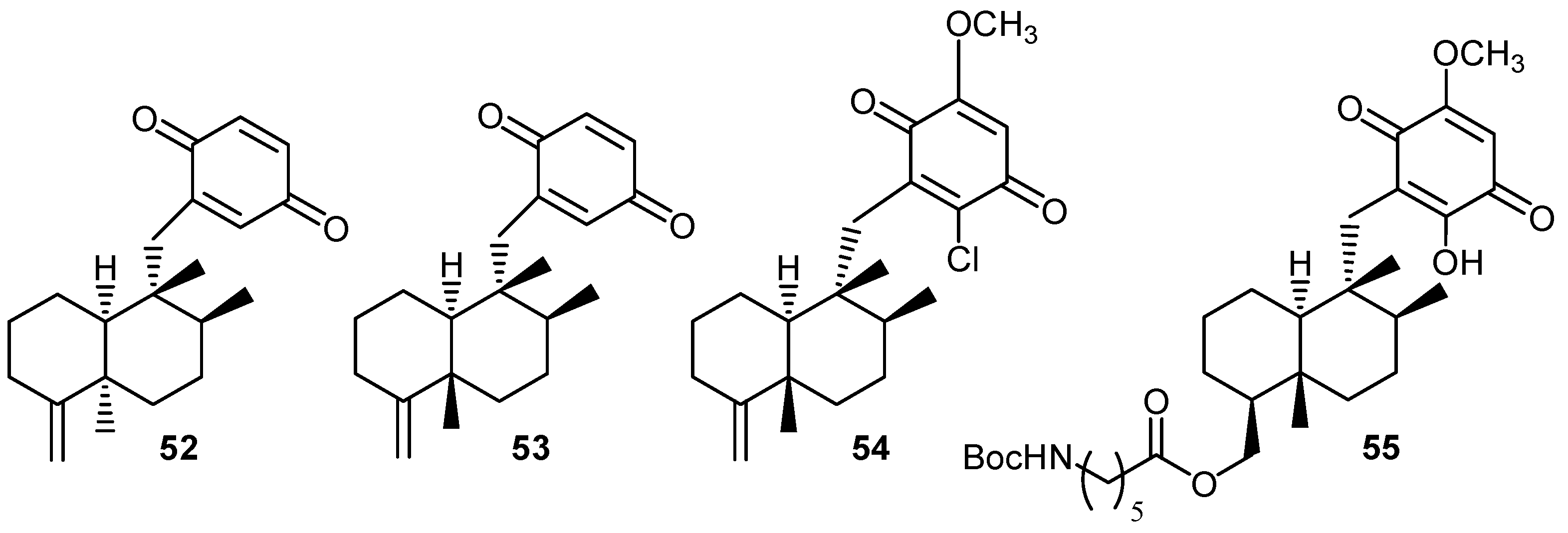
| Compound | Krel [Mg2+]/mol dm-3 | Ep/2, V [Mg2+]/mol dm-3 | |||
|---|---|---|---|---|---|
| 0 | 1.0×10-2 | 1.0×10-1 | 0 | 1.0×10-2 | |
| 2 | 1.4 | 5.1 | 5.4 | -0.56 | -0.23 |
| 20 | 1.6 | 5.0 | 5.3 | -0.62 | -0.34 |
| 33 | 4.2 | 8.9 | 9.0 | -0.43 | -0.20 |
| 34 | 5.9 | 11.8 | 14.3 | -0.56 | -0.36 |
| 35 | 2.4 | 5.6 | 6.4 | -0.58 | -0.22 |
| 36 | 2.3 | 5.9 | 6.5 | -0.57 | -0.34 |
| 37 | 3.9 | 4.6 | 5.0 | -0.38 | -0.20 |
| 38 | 7.2 | 7.8 | 7.8 | -0.18 | – |

| Surfactant | [Mg2+] (mol dm-3) | krel | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 20 | 33 | 34 | 35 | 36 | 37 | 38 | |||||||||
| CTAB | 0 | 3.1* | 3.1 | 6.0 | 9.2 | 1.9 | 2.8 | 4.1 | 7.9 | |||||||
| CTAB | 1.0×10-2 | 5.2§ | 5.0 | 7.1 | 13.8 | 2.7 | 3.5 | 4.4 | 7.9 | |||||||
| CTAB | 1.0×10-1 | 5.2 | 5.2 | 7.0 | 14.0 | 3.0 | 3.5 | 4.6 | 8.1 | |||||||
| SDS | 0 | 1.0†b | 1.4 | 2.5 | 4.8 | 1.9 | 2.0 | 2.9 | 6.6 | |||||||
| SDS | 1.0×10-2 | 1.1‡ | 1.6 | 2.6 | 5.0 | 2.1 | 2.1 | 3.1 | 6.6 | |||||||
| SDS | 1.0×10-1 | 1.1 | 1.7 | 2.7 | 5.1 | 2.1 | 2.2 | 3.1 | 6.7 | |||||||
| TWEEN 80 | 0 | 1.3 | 2.4 | 4.1 | 4.9 | 2.1 | 2.7 | 4.2 | 6.1 | |||||||
| TWEEN 80 | 1.0×10-2 | 1.9 | 3.4 | 4.6 | 5.1 | 2.4 | 2.8 | 4.4 | 6.4 | |||||||
| TWEEN 80 | 1.0×10-1 | 2.1 | 4.0 | 4.6 | 5.2 | 2.8 | 3.4 | 4.5 | 6.7 | |||||||


Arylation of nucleophiles

Tubulin assembly inhibition
Protein kinase inhibition

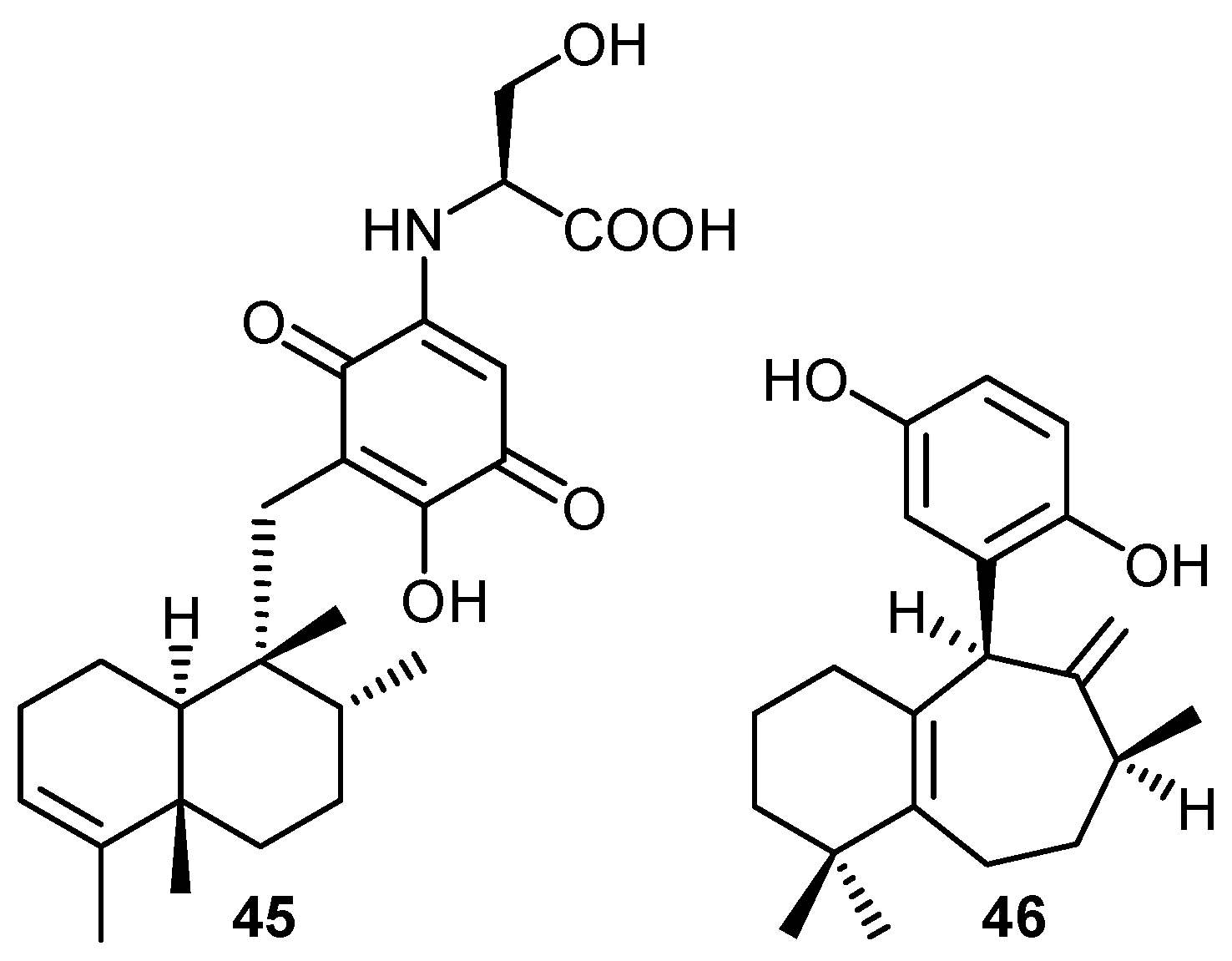
iii) Antiviral activity

iv) Antiinflammatory activity

v) Golgi-disturbing activity


7. Marine pharmacology: perspectives




Acknowledgements
References and Notes
- Berquist, P.R. Sponges; Hutchinson: London, 1978. [Google Scholar]
- Simpson, T.L. The cell biology of sponges; Springer: New York, 1984. [Google Scholar]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2002, 19, 1–48. [Google Scholar] [PubMed]and earlier reviews in this series.
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2005, 22, 15–61. [Google Scholar] [CrossRef] [PubMed]and earlier reviews in this series.
- Faulkner, D.J. Marine Pharmacology. A. van Leeuwenhoek 2000, 77, 135–145. [Google Scholar] [CrossRef]
- Minale, L.; Riccio, R.; Sodano, G. Avarol, a novel sesquiterpenoid hydroquinone with a rearranged drimane skeleton from the sponge Disidea avara. Tetrahedron Lett. 1974, 3401–3404. [Google Scholar] [CrossRef]
- Luibrand, R.T.; Erdman, T.R.; Vollmer, J.J.; Scheur, P.J.; Finer, J.; Clardy, J.C. Ilimaquinone, a sesquiterpenoid quinone from marine sponge. Tetrahedron 1979, 35, 609–612. [Google Scholar] [CrossRef]
- Capon, R.J.; McLeod, J.K. A Revision of the Absolute Stereochemistry of Ilimaquinone. J. Org. Chem. 1987, 52, 5059–5060. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, R.T.; Warren, R.G.; Wells, R.J.; Blount, J.F. New quinones from a dictyoceratid sponge. Aust. J. Chem. 1978, 31, 2685–2697. [Google Scholar] [CrossRef]
- Capon, R.J. The acid-catalyzed rearrangement and absolute stereochemistry of isospongiaquinone. J. Nat. Prod. 1990, 53, 753–756. [Google Scholar] [CrossRef]
- Kobayashi, J.; Madono, T.; Shigemori, H. Nakijiquinone C and D, New Sesquiterpenoid Quinones with a Hydroxy Amino Acid Residue from a Marine Sponge Inhibiting c-erbB-2 Kinase. Tetrahedron 1995, 51, 10867–10874. [Google Scholar] [CrossRef]
- de Guzman, F.; Cop, B.R.; Mayne, C.L.; Concepcion, G.P.; Mangalindan, G.C.; Barrows, L.R.; Ireland, C.M. Bolinaquinone: A Novel Cytotoxic Sesquiterpene Hydroxyquinone from a Philippine Dysidea Sponge. J. Org. Chem. 1998, 63, 8042–8044. [Google Scholar] [CrossRef]
- De Rosa, S.; Minale, L.; Riccio, R.; Sodano, G. The absolute configuration of avarol, a rearranged sesquiterpenoid hydroquinone from a marine sponge. J. Chem. Soc. Perkin Trans.1 1976, 1408–1414. [Google Scholar] [CrossRef]
- Cozzolino, R.; De Giulio, A.; De Rosa, S.; Strazzullo, G.; Gašić, M.J.; Sladić, D.; Zlatović, M. Biological activities of avarol derivatives, 1. amino derivatives. J. Nat. Prod. 1990, 53, 699–702. [Google Scholar] [CrossRef]
- Trifunović, I.; Sladić, D.; Dogović, N.; Gašić, M.J. Carbon-13 chemical shift assignment in substituted 1,4-cyclohexadienones. J. Serb. Chem. Soc. 1987, 52, 559–563. [Google Scholar]
- Sarma, A.S.; Chattopadhyay, P. Synthetic Studies of trans-Clerodane Diterpenoids and Congeners: Stereocontrolled Total Synthesis of (±)-Avarol. J. Org. Chem. 1982, 47, 1727–1731. [Google Scholar] [CrossRef]
- Capon, R.J. Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier Science: Amsterdam, 1995; Vol. 15, pp. 289–326. [Google Scholar]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Makarchenko, A.E. Determination of the Absolute Stereochemistry of Cyclosmenospongine E. J. Nat. Prod. 2003, 66, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Giannini, C.; Debitus, C.; Posadaas, I.; Paya, M.; D Auria, M.V. Dysidotronic acid, a new and selective human phospholipase A2 inhibitor from the sponge Dysidea sp. Tetrahedron Lett. 2000, 41, 3257–3260. [Google Scholar] [CrossRef]
- Patai, S.; Rapaport, Z. (Eds.) The Chemistry of the Quinoid Compounds; Wiley-Interscience: New York, 1988.
- Eberson, L. Electron Transfer Reactions in Organic Chemistry; Springer Verlag: Berlin, 1987. [Google Scholar]
- Kutyrev, A.A. Nucleophilic reactions of quinones. Tetrahedron 1991, 47, 8043–8065. [Google Scholar] [CrossRef]
- Verhoeven, J.W.; van Gerresheim, W.; F. M. Martens, F.M.; van der Kerk, S.M. Mechanism and transition-state structure of hydride-transfer reactions mediated by NAD(P)H-models. Tetrahedron 1986, 42, 975–992. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide Dismutases. Ann. Rev. Biochem. 1975, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar] [CrossRef] [PubMed]and references therein
- O'Brien, P.J. Molecular mechanisms of quinone cytotoxicity. Chem.-Biol. Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Wong, C. L.; Kochi, J. K. Unified view of Marcus electron transfer and Mulliken charge transfer theories in organometallic chemistry. Steric effects in alkylmetals as quantitative probes for outer-sphere and inner-sphere mechanisms. J. Am. Chem. Soc. 1980, 102, 2928–2939. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Koumitsu, S.; Hironaka, K.; Tanaka, T. Energetic Comparison between Photoinduced Electron-Transfer Reactions from NADH Model Compounds to Organic and Inorganic Oxidants and Hydride-Transfer Reactions from NADH Model Compounds to p-Benzoquinone Derivatives. J. Am. Chem. Soc. 1987, 109, 305–316. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Fujii, Y.; Suenobu, T. Metal Ion-Catalyzed Cycloaddition vs Hydride Transfer Reactions of NADH Analogues with p-Benzoquinones. J. Am. Chem. Soc. 2001, 123, 10191–10199. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Ishikawa, M.; Tanaka, T. Acid-catalyzed electron-transfer processes in hydride- transfer reactions from 10-methylacridan to p-benzoquinone derivatives. Chem. Lett. 1989, 1227–1230. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fukuzumi, S. Primary Kinetic Isotope Effects on Acid-catalysed Reduction of p-Benzoquinone Derivatives by an Acid-stable NADH Analogue. J. Chem. Soc. Faraday Trans. 1990, 86, 3531–3536. [Google Scholar] [CrossRef]
- Abeles, R.H.; Hutton, R.F.; Westheimer, F.H. The Reduction of Thioketones by a Model for a Coenzyme. J. Am. Chem. Soc. 1957, 79, 712–716. [Google Scholar] [CrossRef]
- Carlson, B.W.; Miller, L.L. Mechanism of the Oxidation of NADH by Quinones. Energetics of One-Electron and Hydride Routes. J. Am. Chem. Soc. 1985, 107, 479–485. [Google Scholar] [CrossRef]
- Miller, L.; Valentine, J.R. On the Electron-Proton-Electron Mechanism for 1-Benzyl-1,4- dihydronicotinamide Oxidations. J. Am. Chem. Soc. 1988, 110, 3982–3989. [Google Scholar] [CrossRef]
- Coleman, C.J.; Rose, J.G.; Murray, C.J. General acid catalysis of the reduction of p- benzoquinone by an NADH analog. Evidence for concerted hydride and hydron transfer. J. Am. Chem. Soc. 1992, 114, 9755–9762. [Google Scholar] [CrossRef]
- Spyroudis, S. Hydroxyquinones: Synthesis and Reactivity. Molecules 2000, 5, 1291–1330. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.L.; Johnston, J.N. Use of the Vicinal Element Effect for Regiochemical Control of Quinone Substitutions and Its Implication for Convergent Mitomycin Construction. Org. Lett. 2001, 3, 3695–3697. [Google Scholar] [CrossRef] [PubMed]
- Božić, T.; Sladić, D.; Zlatović, M.; Novaković, I.; Trifunović, S.; Gašić, M.J. Regioselectivity of conjugate additions to monoalkyl-1,4-benzoquinones. J. Serb. Chem. Soc. 2002, 67, 547–551. [Google Scholar] [CrossRef]
- Šolaja, B.A.; Milić, D.R.; Gašić, M.J. A Novel m-CPBA Oxidation: p-Quinols and Epoxyquinols from Phenols. Tetrahedron Lett. 1996, 37, 3765–3768. [Google Scholar] [CrossRef]
- Milić, D.R.; Gašić, M.J.; Muster, W.; Csanádi, J.J.; Šolaja, B.A. The Synthesis and Biological Evaluation of A-Ring Substituted Steroidal p-Quinones. Tetrahedron 1997, 53, 14073–14084. [Google Scholar] [CrossRef]
- Milić, D.R.; Kapor, A.; Markov, B.; Ribar, B.; Strümpel, M.; Juranić, Z.; Gašić, M.J.; Šolaja, B.A. X-Ray Crystal Structure of 10β-Hydroxy-4β,5β-epoxyestr-1-en-3,17-dione and Antitumor Activity of its Congeners. Molecules 1999, 4, 338–352. [Google Scholar] [CrossRef]
- Milić, D.R.; Šolaja, B.A.; Došen-Mićović, Lj.; Ribar, B.; Kapor, A.; Sladić, D.; Gašić, M.J. Structure and reactivity of steroidal quinones. J. Serb. Chem. Soc. 1997, 62, 755–768. [Google Scholar]
- Raber, D.J.; Rodriguez, W. Conformational Properties of Oxidation-Reduction Cofactors. J. Am. Chem. Soc. 1985, 107, 4146–4147. [Google Scholar] [CrossRef]
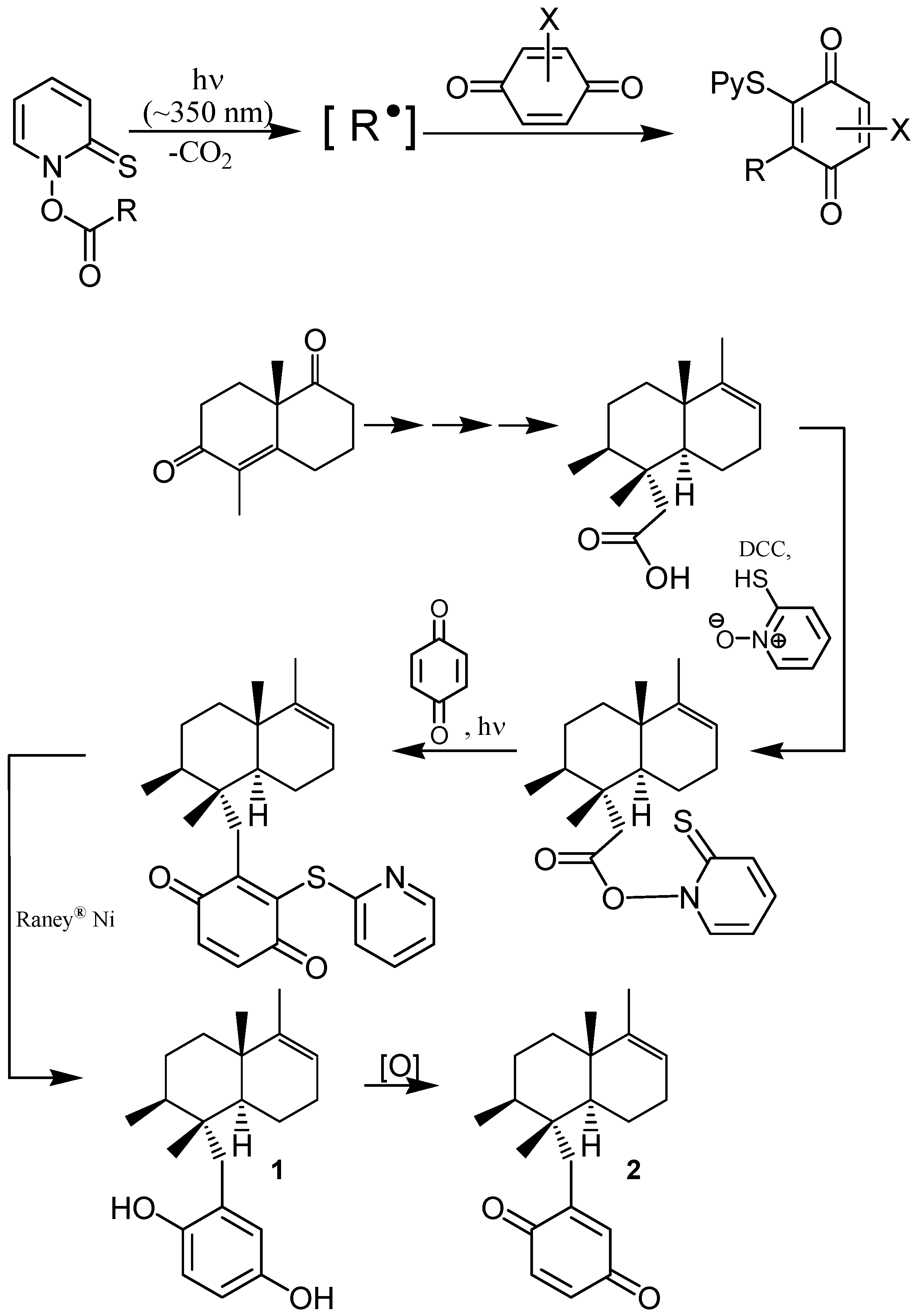
- Ling, T.; Poupon, E.; Rueden, E.J.; Kim, S.H.; Theodorakis, E.A. Unified Synthesis of Quinone Sesquiterpenes Based on a Radical Decarboxylation and Quinone Addition Reaction. J. Am. Chem. Soc. 2002, 124, 12261–12267. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R. The Invention of Chemical-Reactions of Relevance to the Chemistry of Natural Products. Pure Appl. Chem. 1994, 66, 1943–1954. [Google Scholar] [CrossRef]
- Ravi, B.N.; P. Perzanowski, P. H.; Ross, R.; Erdman, T.R.; Scheuer, P.J. Recent research in marine natural products: the puupehenones. Pure Appl. Chem. 1979, 51, 1893–1900. [Google Scholar] [CrossRef]
- Bourguet-Kondracki, M-L.; Lacombe, F.; Guyot, M. Methanol Adduct of Puupehenone, a Biologically Active Derivative from the Marine Sponge Hyrtios sp. J. Nat. Prod. 1999, 62, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, K.A.; Bartyzel, P.; Shen, X.; Perry, T.L.; Zjawiony, J.K.; Hamann, M.T. Marine Natural Products as Antituberculosis Agents. Tetrahedron 2000, 56, 949–953. [Google Scholar] [CrossRef]
- Shen, Y-C.; Hsieh, P-W. New Sesquiterpene Hydroquinones from a Taiwanese Marine Sponge Polyfibrospongia australis. J. Nat. Prod. 1997, 60, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.; Quiñoá, E.; Riguera, R.; Peters, B.M.; Abrell, L.M.; Crews, P. The structures and stereochemistry of cytotoxic sesquiterpene quinones from Dactylospongia elegans. Tetrahedron 1992, 48, 6667–6680. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Zahn, R.K.; Gašić, M.J.; Dogović, N.; Maidhof, A.; Becker, C.; Diehl-Seifert, B.; Eich, E. Avarol, a cytostatically active compound from the marine sponge Dysidea avara. Comp. Biochem. Physiol. 1985, 80C, 47–52. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Maidhof, A.; Zahn, R.K.; Schröder, H.C.; Gašić, M.J.; Heidemann, D.; Bernd, A.; Kurelec, B.; Eich, E.; Seibert, G. Potent Antileukemic Activity of the Novel Cytostatic Agent Avarone and Its Analogues in vitro and in vivo. Cancer Res. 1985, 45, 4822–4826. [Google Scholar] [PubMed]
- Hirsch, S.; Rudi, A.; Kashman, Y.; Loya, Y. New avarone and avarol derivatives from the marine sponge Dysidea cinerea. J. Nat. Prod. 1991, 54, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Lakshmi, V.; Powell, D.R.; Van der Helm, D. Arenarol and arenarone: sesquiterpenoids with rearranged drimane skeletons from the marine sponge Dysidea arenaria. J. Org. Chem. 1984, 49, 241–244. [Google Scholar] [CrossRef]
- Del Corral, J.M.M.; Gordaliza, M.; Castro, M.A.; Mahiques, M.M.; Chamorro, P.; Molinari, A.; García-Grávalos, M.D.; Broughton, H.B.; San Feliciano, A. New Selective Cytotoxic Diterpenylquinones and Diterpenylhydroquinones. J. Med. Chem. 2001, 44, 1257–1267. [Google Scholar] [CrossRef]
- De Giulio, A.; De Rosa, S.; Strazzullo, G.; Diliberto, L.; Obino, P.; Marongiu, M.E.; Pani, A.; La Colla, P. Synthesis and evaluation of cytostatic and antiviral activities of 3' and 4'-avarone derivatives. Antivir. Chem. Chemother. 1991, 2, 223–227. [Google Scholar] [CrossRef]
- Seibert, G.; Raether, W.; Dogović, N.; Gašić, M.J.; Zahn, R.K.; Müller, W.E.G. Antibacterial and antifungal activity of avarone and avarol. Zbl. Bakt. Hyg. 1985, 260A, 379–386. [Google Scholar] [CrossRef]
- Dorta, E.; Cueto, M.; Brito, I.; Darias, J. New Terpenoids from the Brown Alga Stypopodium zonale. J. Nat. Prod. 2002, 65, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y-C.; Chen, C-Y.; Kuo, Y-H. New Sesquiterpene Hydroquinones from a Taiwanese Marine Sponge, Hippospongia metachromia. J. Nat. Prod. 2001, 64, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Hardt, I.H.; Jensen, P.R.; Fenical, W. Neomarinone, and new cytotoxic marinone derivatives, produced by a marine filamentous bacterium (actinomycetales). Tetrahedron Lett. 2000, 41, 2073–2076. [Google Scholar] [CrossRef]
- Warabi, K.; McHardy, L.M.; Matainaho, L.; Van Soest, R.; Roskelley, C.D.; Roberge, M.; Andersen, R.J. Strongylophorine-26, a New Meroterpenoid Isolated from the Marine Sponge Petrosia (Strongylophora) corticata That Exhibits Anti-invasion Activity. J. Nat. Prod. 2004, 67, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Herrador, M.M.; Chahboun, R.; Galera, P. Synthesis and Antitumoral Activities of Marine ent-Chromazonarol and Related Compounds. Bioorg. Med. Chem. 1999, 9, 2325–2328. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Herrador, M.M.; Valdivia, M.V.; Chahboun, R. Synthesis of Monoterpenic Analogues of Puupehenone and Puupehedione. Tetrahedron Lett. 1998, 39, 2425–2428. [Google Scholar] [CrossRef]
- Henry, R.T.; Wallace, B.K. Different Mechanisms of Cell Killing by Redox Cycling and Arylating Quinones. Arch. Toxicol. 1996, 70, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Guilivi, C.; Cadenas, E. One- and two-electron reduction of 2-methyl-1,4-naphthoquinone bioreductive alkylating agents: kinetic studies, free-radical production, thiol oxidation and DNA- strand-break formation. Biochem. J. 1994, 301, 21–30. [Google Scholar] [CrossRef]
- Ernster, L. DT diaphorase: a historical review. Chem. Scripta 1987, 27A, 1–13. [Google Scholar]
- Maruyama, A.; Kumagai, Y.; Morikawa, K.; Taguchi, K.; Hayashi, H.; Ohta, T. Oxidative-stress- inducible qorA encodes an NADPH-dependent quinone oxidoreductase catalysing a one-electron reduction in Staphylococcus aureus. Microbiology 2003, 149, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.A. On the application of electron paramagnetic resonance in the study of naturally occurring quinones and quinols. Spectrochim. Acta A 2002, 58, 1257–1270. [Google Scholar] [CrossRef]
- Gašić, M.J. Biologically active compounds from marine sponges: an approach to chemical and biochemical characterization of the avarol/avarone redox couple. J. Serb. Chem. Soc. 1988, 53, 229–249. [Google Scholar]
- Müller, W.E.G.; Sladić, D.; Zahn, R.K.; Bässler, K-H.; Dogović, N.; Gerner, H.; Gašić, M.J.; Schröder, Heinz C. Avarol-induced DNA strand breakage in vitro and in Friend erythroleukemia cells. Cancer Res. 1987, 47, 6565–6571. [Google Scholar]
- Schröder, H.C.; Wenger, R.; Gerner, H.; Reuter, P.; Kuchino, Y.; Sladić, D.; Müller, W.E.G. Suppression of the Modulatory Effects of the Antileukemic an the Anti-Human Immunodeficiency Virus Compound Avarol on Gene Expression by Tryptophan. Cancer Res. 1989, 49, 2069–2076. [Google Scholar] [PubMed]
- Sladić, D.; Gašić, M.J. Effects of iron(II) compounds on the amount of DNA damage in Friend erythroleukemia cells induced by avarol. Role of hydroxyl radicals. J. Serb. Chem. Soc. 1994, 59, 915–920. [Google Scholar]
- Batke, E.; Schröder, H.C.; Prellwitz, W.; Maidhof, A.; Eich, E.; Zahn, R.K.; Gašić, M.J.; Müller, W.E.G. Influence of the antileukemic agents avarone and avarol on superoxide dismutase in vitro as well as in vivo (L5178y mouse lymphoma cells). Biochem. Arch. 1987, 3, 275–284. [Google Scholar]
- Batke, E.; Ogura, R.; Vaupel, P.; Hummel, K.; Kallinowski, F.; Gašić, M.J.; Schröder, H.C.; Müller, W.E.G. Action of the antileukemic and anti-HTLV-III (anti-HIV) agent avarol on the levels of superoxide dismutases and glutathione peroxidase activities in L5178y mouse lymphoma cells. Cell. Biochem. Funct. 1987, 6, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Gašić, M.J.; Sladić, D.; Tabaković, I.; Davidović, A. The Avarol-Avarone Redox Behaviour in Acetonitrile. Croat. Chem. Acta 1985, 58, 531–536. [Google Scholar]
- Tabaković, I.; Davidović, A.; Müller, W.E.G.; Zahn, R.K.; Sladić, D.; Dogović, N.; Gašić, M.J. Electrochemical reactivity of biologically active quinone/hydroquinone sesquiterpenoids on glassy carbon electrodes. Bioelectrochem. Bioener. 1987, 17, 567–577. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Diehl-Seifert, B.; Sobel, C.; Bechtold, A.; Kljajić, Z.; Dorn, A. Sponge secondary metabolites: biochemical and ultrastructural localization of the antimitotic agent avarol in Dysidea avara. J. Histochem. Cytochem. 1986, 34, 1687–1690. [Google Scholar]
- Dogović, N.; Sladić, D.; Gašić, M.J.; Tabaković, I.; Davidović, A.; Gunić, E. Reactivity of avarone and related p-benzoquinones with NADH model compounds. Gazz. Chim. Ital. 1991, 121, 63–66. [Google Scholar]
- Davidović, A.; Tabaković, I.; Sladić, D.; Dogović, N.; Gašić, M.J. Mechanism of oxidation of 1- benzyl-1,4-dihydronicotinamide by the biologically active p-benzoquinone derivative, avarone, in a cationic micellar medium. Bioelectrochem. Bioener. 1991, 26, 457–468. [Google Scholar] [CrossRef]
- Zlatović, M.; Sladić, D.; Gašić, M.J. The kinetics of the reduction of the lipophilic quinone avarone by n-alkyl-1,4-dihydronicotinamides of various lipophilicities. J. Serb. Chem. Soc. 1999, 64, 647–654. [Google Scholar]
- Okubo, T.; Nagai, F.; Ushiyama, K.; Kano, I. Contribution of oxygen radicals to DNA cleavage by quinone compounds derived from phenolic antioxidants, tert-butylhydroquinone and 2,5-di- tert-butylhydroquinone. Toxicol. Lett. 1997, 90, 11–18. [Google Scholar] [CrossRef]
- Swersey, J.C.; Barrows, L.R.; Ireland, C.M. Mamanuthaquinone: an antimicrobial and cytotoxic metabolite of Fasciospongia sp. Tetrahedron Lett. 1991, 46, 6687–6690. [Google Scholar] [CrossRef]
- Belisario, M.A.; Pecce, R.; Maturo, M.; De Rosa, S. Arylation of sulfhydryl groups in vitro by the naturally occurring sesquiterpenoid benzoquinone avarone. Toxicology 1994, 86, 89–108. [Google Scholar] [CrossRef]
- Belisario, M.A.; Maturo, M.; Avagnale, G.; De Rosa, S.; Scopacasa, F.; De Caterina, M. In vitro Effect of Avarone and Avarol, a Quinone/Hydroquinone Couple of Marine Origin, on Plattelet Aggregation. Pharmacol. Toxicol. 1996, 79, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Novaković, I.; Vujčić, Z.; Božić, T.; Božić, N.; Milosavić, N.; Sladić, D. Chemical modification of β-lactoglobulin by quinones. J. Serb.Chem.Soc. 2003, 68, 243–248. [Google Scholar] [CrossRef]
- Sladić, D.; Novaković, I.; Vujčić, Z.; Božić, T.; Božić, N.; Milić, D.; Šolaja, B.; Gašić, M.J. Protein covalent modification by biologically active quinones. J. Serb. Chem. Soc. 2004, 69, 901–907. [Google Scholar] [CrossRef]
- Zjawiony, J.K.; Bartyzel, P.; Hamann, M.T. Chemistry of Puupehenone: 1,6-Conjugate Addition to Its Quinone-Methide System. J. Nat. Prod. 1998, 61, 1502. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Dogović, N.; Zahn, R.K.; Maidhof, A.; Diehl-Seifert, B.; Becker, C.; Sachsse, W.; Gašić, M.J.; Schröder, H.C. Inhibition of mitosis by avarol, a natural product isolated from the sponge Dysidea avara. Bas. Appl. Histochem. 1985, 29, 321–330. [Google Scholar]
- Sandoval, I.V.; Weber, K. Polymerization of tubulin in the presence of colchicine or podophyllotoxin. Formation of a ribbon structure induced by guanylyl-5'-methylene diphosphonate. J. Mol. Biol. 1979, 134, 159–172. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Tolcher, A.W. Cancer: Principles and Practice of Oncology; DeVita, V.T., Jr., Hellman, S., Rosenberg, S.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2001; Vol. 1, pp. 431–447. [Google Scholar]
- Cohen, P. Protein kinases - the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Bridges, A.J. Chemical Inhibitors of Protein Kinases. Chem. Rev. 2001, 101, 2541–2571. [Google Scholar] [CrossRef] [PubMed]
- Wessels, M.; König, G.M.; Wright, A.D. A New Tyrosine Kinase Inhibitor from the Marine Brown Alga Stypopodium zonale. J. Nat. Prod. 1999, 62, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.-D.; Leung, D.; Sanghara, J.; Daley, D.; van Soest, R.; Andersen, R.J. Isoarenarol, A New Protein Kinase Inhibitor from the Marine Sponge Dysidea arenaria. Pharm. Biol. 2003, 41, 223–225. [Google Scholar] [CrossRef]
- Alvi, K.A.; Diaz, M.C.; Crews, P.; Slate, D.L.; Lee, R.H.; Moretti, R. Evaluation of New Sesquiterpene Quinones from Two Dysidea Sponge Species as Inhibitors of Protein Tyrosine Kinase. J. Org. Chem. 1992, 57, 6604–6607. [Google Scholar] [CrossRef]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.; Waldmann, H. Total Synthesis and Biological Evaluation of the Nakijiquinones. J. Am. Chem. Soc. 2001, 123, 11586–11593. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.D.; Freyer, A.J.; Killmer, L.; Offen, P.; Carte, B.; Jurewicz, A.J.; Johnson, R.K. Frondosins, Five New Sesquiterpene Hydroquinone Derivatives with Novel Skeletons from the Sponge Dysidea frondosa: Inhibitors of Interleukin-8 Receptors. Tetrahedron 1997, 53, 5047–5060. [Google Scholar] [CrossRef]
- Sarin, P.S.; Sun, D.; Thornton, A.; Müller, W.E.G. Inhibition of replication of the etiological agent of acquired immune deficiency syndrome (human T-lymphotropic retrovirus/lymphadenopathy-associated virus) by avarol and avarone. J. Natl. Cancer Inst. 1987, 78, 663–666. [Google Scholar] [PubMed]
- Loya, S.; Hizi, A. The inhibition of human immunodeficiency virus type 1 reverse transcriptase by avarol and avarone derivatives. FEBS Lett. 1990, 269, 131–134. [Google Scholar] [CrossRef]
- Loya, S.; Tal, R.; Kashman, Y.; Hizi, A. Illimaquinone, a selective inhibitor of the RNase H activity of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1990, 34, 2009–2012. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Gao, Z.; Thomas, S.J.; Hecht, S.M.; Lazo, J.S.; Kingston, D.G.I. Marine Sesquiterpenoids that Inhibit the Lyase Activity of DNA Polymerase β. J. Nat. Prod. 2004, 67, 1716–1718. [Google Scholar] [CrossRef] [PubMed]
- Kuchino, Y.; Nishimura, S.; Schröder, H.C.; Rottmann, M.; Müller, W.E.G. Selective inhibition of formation of suppressor glutamine tRNA in Moloney murine leukemia virus-infected NIH- 3T3 cells by Avarol. Virology 1988, 165, 518–526. [Google Scholar] [CrossRef]
- Loya, S.; Bakhanashvili, M.; Kashman, Y.; Hizi, A. Peyssonols A and B, two novel inhibitors of the reverse transcriptases of human immunodeficiency virus types 1 and 2. Arch. Biochem. Biophys. 1995, 316, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Loya, S.; Rudi, A.; Kashman, Y.; Hizi, A. Mode of inhibition of HIV reverse transcriptase by 2- hexaprenylhydroquinone, a novel general inhibitor of RNA- and DNA-directed DNA polymerases. Biochem. J. 1997, 324, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Tziveleka, L.-A.; Vagias, C.; Roussis, V. Natural products with anti-HIV activity from marine organisms. Curr. Top. Med. Chem. 2003, 3, 1512–1535. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.C.; Begin, M.E.; Kloecking, R.; Matthes, E.; Sarma, S.S.; Gašić, M.J.; Mueller, W.E.G. Avarol restores the altered prostaglandin and leukotriene metabolism in monocytes infected with human immunodeficiency virus type 1. Virus Research 1991, 21, 213–23. [Google Scholar] [CrossRef]
- Ferrándiz, M.L.; Sanz, M.J.; Bustos, G.; Payá, M.; Alcaraz, M.J.; De Rosa, S. Avarol and avarone, two new anti-inflammatory agents of marine origin. Eur. J. Pharmacol. 1994, 253, 75–82. [Google Scholar] [CrossRef]
- Amigó, M.; Terencio, M.C.; Mitova, M.; Iodice, C.; Payá, M.; De Rosa, S. Potential Antipsoriatic Avarol Derivatives as Antioxidants and Inhibitors of PGE2 Generation and Proliferation in the HaCaT Cell Line. J. Nat. Prod. 2004, 67, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Giannini, C.; D’Auria, M.V.; Payá, M. Modulatory Effect of Bolinaquinone, a Marine Sesquiterpenoid, on Acute and Chronic Inflammatory Processes. J. Pharmacol. Exp. Ther. 2003, 304, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Amagata, T.; Whitman, S.; Johnson, T.A.; Stessman, C.C.; Loo, C.P.; Lobkovsky, E.; Clardy, J.; Crews, P.; Holman, T.R. Exploring Sponge-Derived Terpenoids for Their Potency and Selectivity against 12-Human, 15-Human, and 15-Soybean Lipoxygenases. J. Nat. Prod. 2003, 66, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Gil, B.; Sanz, M.J.; Terencio, M.C.; De Giulio, A.; De Rosa, S.; Alcaraz, M.J.; Payá, M. Effects of marine 2-ployprenyl-1,4-hydroquinones on phospholipase A2 activity and some inflammatory responses. Eur. J. Pharmacol. 1995, 285, 281–288. [Google Scholar] [CrossRef]
- Dinter, A.; Berger, E.G. Golgi-disturbing agents. Histochem. Cell. Biol. 1998, 109, 571–590. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, P.A.; Yucel, J.K.; Veit, B.; Faulkner, D.J.; Deerinck, T.; Soto, G.; Ellisman, M.; Malhotra, V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell 1993, 73, 1079. [Google Scholar] [CrossRef]
- Rangel, H.R.; Dagger, F.; Compagnone, R.S. Antiproliferative effect of illimaquinone on Leishmania mexicana. Cell. Biol. Internat. 1997, 21, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Radeke, H.S.; Digits, C.A.; Casaubon, R.L.; Snapper, M.L. Interactions of (-)-ilimaquinone with methylation enzymes: implications for vesicular-mediated secretion. Chem. Biol. 1999, 6, 639–647. [Google Scholar] [CrossRef]
- Battershill, C.N.; Page, M.J. Sponge aquaculture for drug production. Aquacult. Updates 1996, 16, 5–6. [Google Scholar]
- Adams, C.; Stevely, J.M.; Sweat, D.J. Economic feasibility of small-scale sponge farming in Pohnpei, Federated States of Micronesia. J. World Aquacult. 1995, 26, 132–142. [Google Scholar] [CrossRef]
- Barthel, D.; Theede, H. A new method for the culture of marine sponges and its application for experimental studies. Ophelia 1986, 25, 75–82. [Google Scholar] [CrossRef]
- De Rosa, S. Natural Products in the New Millennium: Prospects and Industrial Application; Rauter, A.P., Palma, F.B., Araujo, M.A., dos Santos, S.P., Eds.; Kluwer Academic Publishers: Dordrecht, 2002; pp. 441–461. [Google Scholar]
- Müller, W.E.G.; Wiens, M.; Batel, R.; Steffen, R.; Borojevic, R.; Custodio, M.R. Established of a primary cell culture from a sponge: primmorphs from Suberites domuncula. Mar. Ecol. Prog. Ser. 1999, 178, 205–219. [Google Scholar] [CrossRef]
- Engel, S.; Jensen, P.R.; Fenical, W. Chemical ecology of marine microbial defense. J. Chem.Ecol. 2002, 28, 1971–1985. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; et al. Microbes from deep ocean sediments, accessing a new antibiotic resource. In International Symposium “Chemistry & Biology of Marine Organisms”, Kolympari, Crete, Greece, Sept. 21- 26, 2003; Crete, 2003. [Google Scholar]
- Fenical, W.; Sethna, K.; Lloyd, G.K. Pharma. News. 2002, 9, 489–494.
- Nicolaou, K.C.; Vourloumis, D.; Winssinger, N.; Baran, P.S. The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century. Angew. Chem. Int. Ed. 2000, 39, 44–122. [Google Scholar] [CrossRef] and ref therein
- Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.; Waldmann, H. Total Synthesis and Biological Evaluation of the Nakijiquinones. J. Am. Chem. Soc. 2001, 47, 11586–11593. [Google Scholar] [CrossRef]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Giannis, A.; Waldmann, H. Natural Product Derived Receptor Tyrosine Kinase Inhibitors: Identification of IGF1R, Tie-2, and VEGFR-3 Inhibitors. Angew. Chem. Int. Ed. 2002, 41, 1174–1178. [Google Scholar] [CrossRef]
- Wender, P.A.; Hilinski, M.K.; Mayweg, A.V.W. Late-State Intermolecular CH Activation for Lead Diversification: A Highly Chemoselective Oxyfunctionalization of the C-9 Position of Potent Bryostatin Analogues. Org. Lett. 2005, 7, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Baryza, J.L.; Bennet, C.E.; Bi, F.C.; Brenner, S.E.; Clarke, M.O.; Horan, J.C.; Kan, C.; Lacôte, E.; Lippa, B.; Nell, P.G.; Turner, T.M. The Practical Synthesis of a Novel and Highly Potent Analogue of Bryostatin. J. Am. Chem. Soc. 2002, 46, 13648–13649. [Google Scholar] [CrossRef] and references therein
- Mooberry, S.L.; Randall-Hlubek, D.A.; Leal, R.M.; Hegde, S.G.; Hubbard, R.D.; Zhang, L.; Wender, P.A. Microtubule-stabilizing agents based on designed laulimalide analogues. Proc. Natl. Acad. Sci. USA 2004, 101, 8803–8808. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar] [PubMed]
- Sample availability: Not applicable
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sladic, D.; Gasic, M.J. Reactivity and Biological Activity of the Marine Sesquiterpene Hydroquinone Avarol and Related Compounds from Sponges of the Order Dictyoceratida. Molecules 2006, 11, 1-33. https://doi.org/10.3390/11010001
Sladic D, Gasic MJ. Reactivity and Biological Activity of the Marine Sesquiterpene Hydroquinone Avarol and Related Compounds from Sponges of the Order Dictyoceratida. Molecules. 2006; 11(1):1-33. https://doi.org/10.3390/11010001
Chicago/Turabian StyleSladic, Dusan, and Miroslav J. Gasic. 2006. "Reactivity and Biological Activity of the Marine Sesquiterpene Hydroquinone Avarol and Related Compounds from Sponges of the Order Dictyoceratida" Molecules 11, no. 1: 1-33. https://doi.org/10.3390/11010001