Improving the Quality of Published Chemical Names with Nomenclature Software

Department of Drug Synthesis, Faculty of Life Sciences, University of Vienna, Althanstrasse 14, 1090 Vienna, Austria
Molecules 2006, 11(11), 915-928; https://doi.org/10.3390/11110915
Submission received: 8 November 2006
/
Revised: 28 November 2006
/
Accepted: 28 November 2006
/
Published: 29 November 2006
Abstract
:This work deals with the use of organic systematic nomenclature in scientific literature, its quality, and computerized methods for its improvement. Criteria for classification of systematic names in terms of quality/correctness are discussed and applied to a sample set of several hundred names extracted from the literature. The same structures are named with three popular state-of-the-art nomenclature programs – AutoNom 2000, ChemDraw 10.0, and ACD/Name 9.0. When comparing the results, all nomenclature tools show a significantly better performance than 'average chemists'. One program allows the generation not only of IUPAC names but also of CAS-like index names that are compared with the officially registered names. The scope and limitations of nomenclature software are discussed and a comparison of the programs' actual capabilities is given.
Introduction
Since the very beginning of chemistry as a scientific discipline, names have been assigned to chemical compounds. Initially, trivial names were used; however, with the dramatic increase in the number of known substances, this naming method became impractical since these unsystematic names – typically derived from some property of the compound or its origin, for example, mandelic acid – did not provide any information about the compound's structure [1,2,3]. Hence, the need for a systematic naming system grew and culminated in the first conference on systematic nomenclature held in Geneva in 1892 and in the foundation of the International Union of Pure and Applied Chemistry (IUPAC) in 1919 [4]. Since then the IUPAC has been publishing nomenclature recommendations that provide the scientific community with general guidelines for naming chemical compounds systematically [5,6,7,8]. These rules may be considered as the basic chemists' language, one that may occasionally be spoken using slightly different dialects – as e.g. by the Chemical Abstracts Service (CAS) or by the Beilstein-Institut. Like any modern language, systematic nomenclature is still under development as rule adoptions and enlargements to accommodate new subjects demonstrate [9,10,11]. Despite this continuing process, nomenclature rules cannot be conclusive – yet their main intention remains the same: the transformation of a chemical structure into a name that can be read, printed, and communicated easily and from which the original structure can be generated [12].
However, this continuing process, as well as the expansion to modern fields of chemistry, complicates the correct application of the nomenclature rules. Even if every scientist received a more or less thorough introduction to basic nomenclature during his education, it would be a moot point if he does handle or even wants to apply the once learned knowledge in practice. Testing this behavior might be an interesting point to investigate, especially, since almost every international chemistry journal commits its authors to the use of systematic nomenclature [13], at least in the experimental section when prepared or isolated compounds have to be meticulously characterized and of course unambiguously named. It certainly is a waste of time and resources when the hard-won results of someone's research become more or less worthless due to misinterpretation or misunderstanding of the chemical names describing the relevant compounds.
When computers became more popular and widely used in the last decades of the past century, several attempts were made to computerize and hence to facilitate the naming procedure. In 1991, about 30 years after Garfield’s pioneering work on nomenclature algorithms [14], the first commercial software package – AutoNom – was released by the German Beilstein-Institut [15,16]. Products from some other companies followed later in the 90's [17], but the use of their programs was limited due to severe restrictions to a quite small number of accepted classes of compounds or due to the generation of not preferred, unnecessarily lengthy names. In the meantime, new and improved versions of nomenclature software have become readily accessible, typically as add-ins or components of structure drawing programs. Currently, the most important of these include: AutoNom [18] (although its parent drawing software, ISIS/Draw is no longer updated and improved by MDL, it is widely used since the full version is available for free), ChemDraw [19], and ACD/Name [20]. Other software products have entered the market in recent years, e.g.: NameIt [21], LexiChem [22] and Nomenclator [23].
Although some of these parent drawing packages (in most cases the older versions) were described in several software reviews [24,25,26,27,28,29], scant attention has been paid to these nomenclature tools [30,31]; furthermore, a scholarly head-to-head comparison has not yet been carried out. Hence, the present work is intended to address this issue by analyzing the software programs' quality and comparing these results with the authors' performance in assigning systematic names manually.
Methods
For this survey, the published articles from the first annual issue of the following four chemical journals (year/publisher) were analyzed: European Journal of Medicinal Chemistry (2005, Elsevier), Heteroatom Chemistry (2004, Wiley), Journal of Organic Chemistry (2005, American Chemical Society), and Monatshefte für Chemie/Chemical Monthly (2004, Springer) [32]. Obviously, thousands of compounds are discussed in these issues and most have been named; clearly it would not be reasonable to check all of these names. On the one hand, this would be extremely time-consuming, and on the other, it does not seem to be appropriate for this analysis, since a single paper could too easily distort the overview; e.g. an article in which a new esterification method is exemplified with hundreds of only slightly different halobenzoic acid ethyl esters (e.g. ethyl 2-chlorobenzoate, ethyl 3-chlorobenzoate, ethyl 4-chlorobenzoate, ethyl 2-bromobenzoate, and so on). To overcome these difficulties and to allow a representative analysis, as far as possible, no more than five compounds per paper were selected. In those cases in which more substances were named, five of them had to be selected manually.
However, whenever this necessary 'data-reduction' was carried out, a selection of 'diverse' compounds was attempted in order to minimize less meaningful clusters of very similarly named compounds (see the above example of the ethyl halobenzoates). At this point it must be stated that it seemed to be difficult or even impossible to find absolutely neutral criteria for selection – molecular 'diversity' [33,34], an essential point, for example, in quantitative structure activity relationship (QSAR) studies, is a challenging problem. Nevertheless, from a statistical point of view this lack of perfect randomness in these specific cases is negligible.
Furthermore, this testing was restricted to 'typical' organics. Thus, isotopically modified compounds, macromolecules (such as proteins, polysaccharides, etc.), organometallics/coordination compounds, radicals, polymers, cyclophanes, fullerenes, and other less usual or complicated classes of compounds were ignored.
The structures of every one of the 303 extracted compounds were manually entered into the nomenclature tool and then named. For this purpose, the most recent versions of the following programs were used – AutoNom 2000 (embedded in the well-known ISIS/Draw 2.5), the 'Struct=Name'-tool of ChemDraw 10.0 [35], and ACD/Name 9.08 (running on ACD/ChemSketch 9.08). It should be mentioned that for the latter two programs only minimal differences with the preceding release were observed (newer versions have become available since the writing of this paper). Whereas the changes in ACD/Name affected only sophisticated areas of nomenclature – the basics seem to be absolutely mastered – in ChemDraw some weaker algorithms were slightly improved to increase the number of supported structural elements.
For ACD/Name, which is the only software module allowing the user to set preferences, the default options were chosen [36]. Furthermore, this software permits name generation according to both IUPAC and CAS rules [37]. At first glance, this extra feature seems to be less useful, but many chemists prefer CAS names, which are similar or identical to the IUPAC ones in most cases (except for the name-inversion). However, the CAS names' advantage is that millions of manually named structures can be looked up in a collective index. These names can sometimes be very helpful for naming compounds similar or analogous to ones previously indexed to the CAS database.
Finally, the published names as well as all the generated ones were manually checked for their correctness and classified as follows:
- No name (N): The software was unable to generate a name for the input structure.
- Unacceptable (X): It is not unambiguously possible to generate the correct structure from the name.
- Unambiguous (U): All other names.
For a better understanding of this procedure, in Figure 1 every 20th structure of the entire data set is shown, together with the (generated) names and their classification according to the described rules.
Figure 1.
Every 20th structure of the data set with the corresponding names.
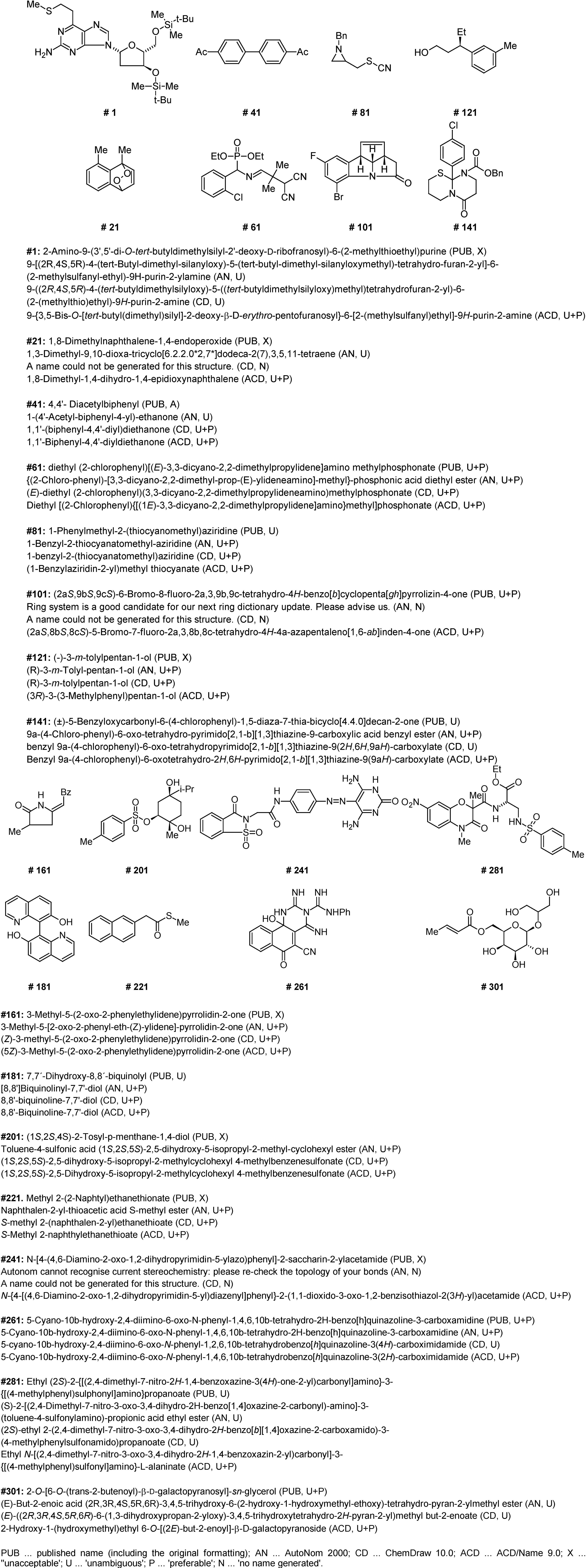
Since the primary goal of systematic nomenclature is to give each compound a label from which the original structure can be perceived, the feasibility of this structure generation process ('unambiguous' names) was regarded as the conditio sine qua non. Nonetheless, it seems to be very desirable to differentiate within this class of names, simply because even when the rules of systematic nomenclature are broken in many ways the name may be unambiguous. For example, both 6‑chloronicotinic acid and 3-hydroxyformyl-1,2,3,4,5,6-hexadehydro-1-azacyclohexan-6-yl chloride unequivocally describe the same structure, although the latter name obviously violates several nomenclature rules. Unfortunately, it turned out that it is easier said than done to 'quantify' a systematic name's quality because a correct name following the specific rules is not necessarily the sole correct name. In the weird example above, while the name may be quite clear, in some other cases only a thin line exists between 'good' and 'bad' names. But where to draw the line? Even restricting it to only IUPAC names does not solve the problem, because these rules often leave a choice, have been changed and improved, or are sometimes contradictory. Presently, the IUPAC is working on a worthwhile project to provide concise rules that would generate a unique name for each structure (the so called 'PIN' – preferred IUPAC name) [8]. Anyway, other name possibilities could be still accepted, but not recommended (compare the different but correct names for Me2CO: acetone, dimethyl ketone, propanone, 2-propanone, propan-2-one). This work has attempted to at least find a way to estimate a 'quality' tendency by setting specifications that consider which names deemed 'unambiguous' are better and which are worse.
The following criteria were regarded to be best fitting for defining a 'preferable' (P) systematic name in this survey: The name is unambiguous, reproducible, and correct in accordance to systematic rules. When rules leave a choice or when different systematic nomenclature systems may be used, all possible names are considered. Exceptions are: lengthy systematic names for amino acids and carbohydrates (e.g. β-d-glucopyranose versus (2R,3R,4S,5S,6R)-6-(hydroxymethyl)tetrahydro-2H-pyran-2,3,4,5-tetrol) as well as needless use of replacement nomenclature or von Baeyer names for (bridged) fused systems (e.g. 5,8-ethanocinnoline versus 3,4-diazatricyclo[6.2.2.02,7]dodeca-1(10),2,4,6,8-pentaene).
Some typical reasons/errors why an 'unambiguous' name was not regarded as 'preferable' include: wrong alphabetical order; principal functional group (suffix) not correctly expressed; wrong/missing stereochemical assignment ('CIP-rules') notwithstanding known/identified stereochemistry; error in expression of degree of saturation ('indicated hydrogen'); wrong order of seniority; absence of a multiplicative name; wrong numbering. The use of italicization, enclosing marks, hyphens, spaces, special characters for formatting in computer software, racemic and relative stereodescriptors, and obvious typos (e.g. ribofranosyl instead of ribofuranosyl) were ignored provided that this did not cause ambiguities.
Separately, the CAS style names generated with ACD/Name (Index) (the only software package providing this feature) were compared with their CAS Registry names (as accessed online via SciFinder Scholar 2006 [38]) and classified as being identical or not.
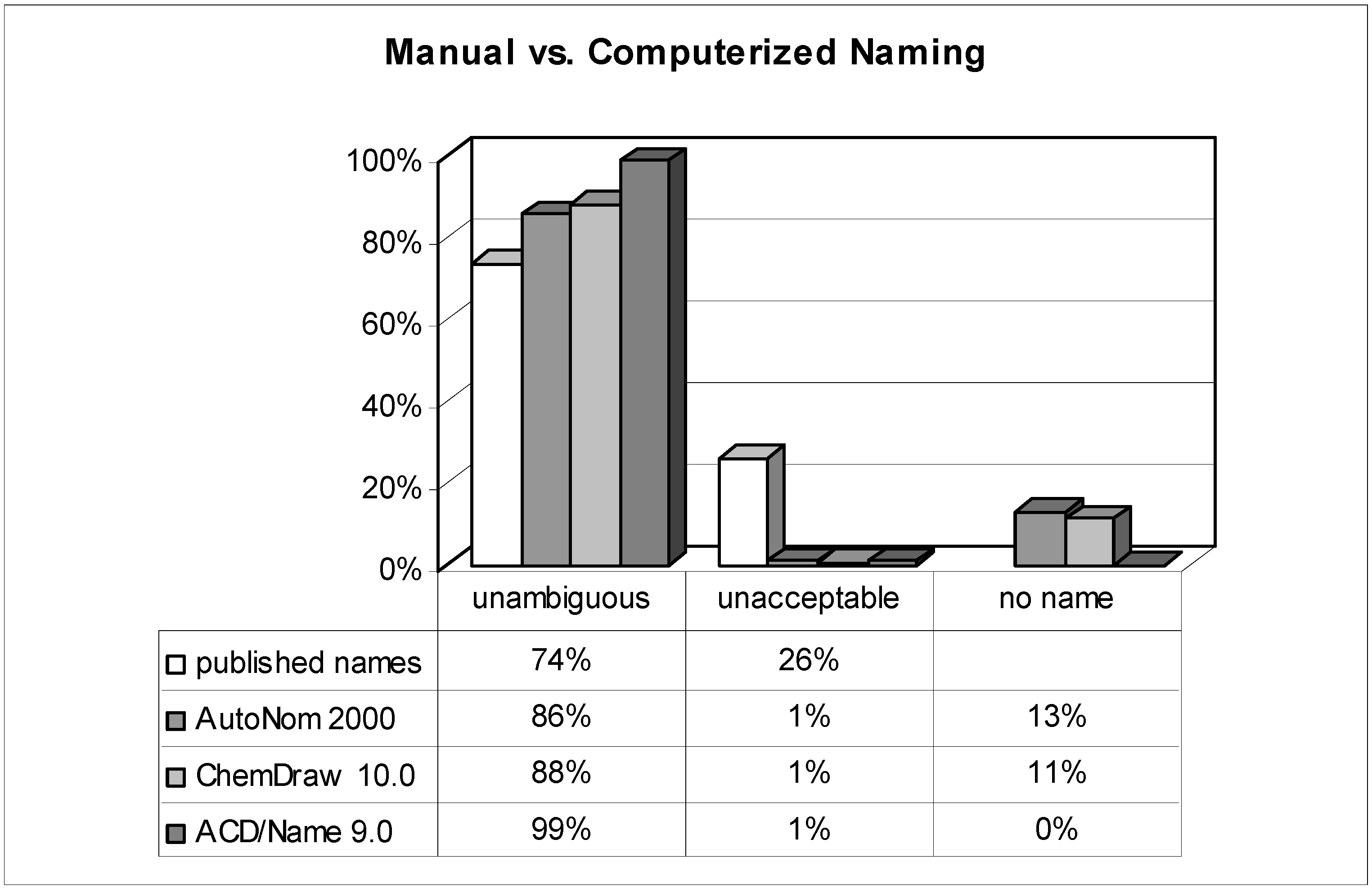
Figure 2.
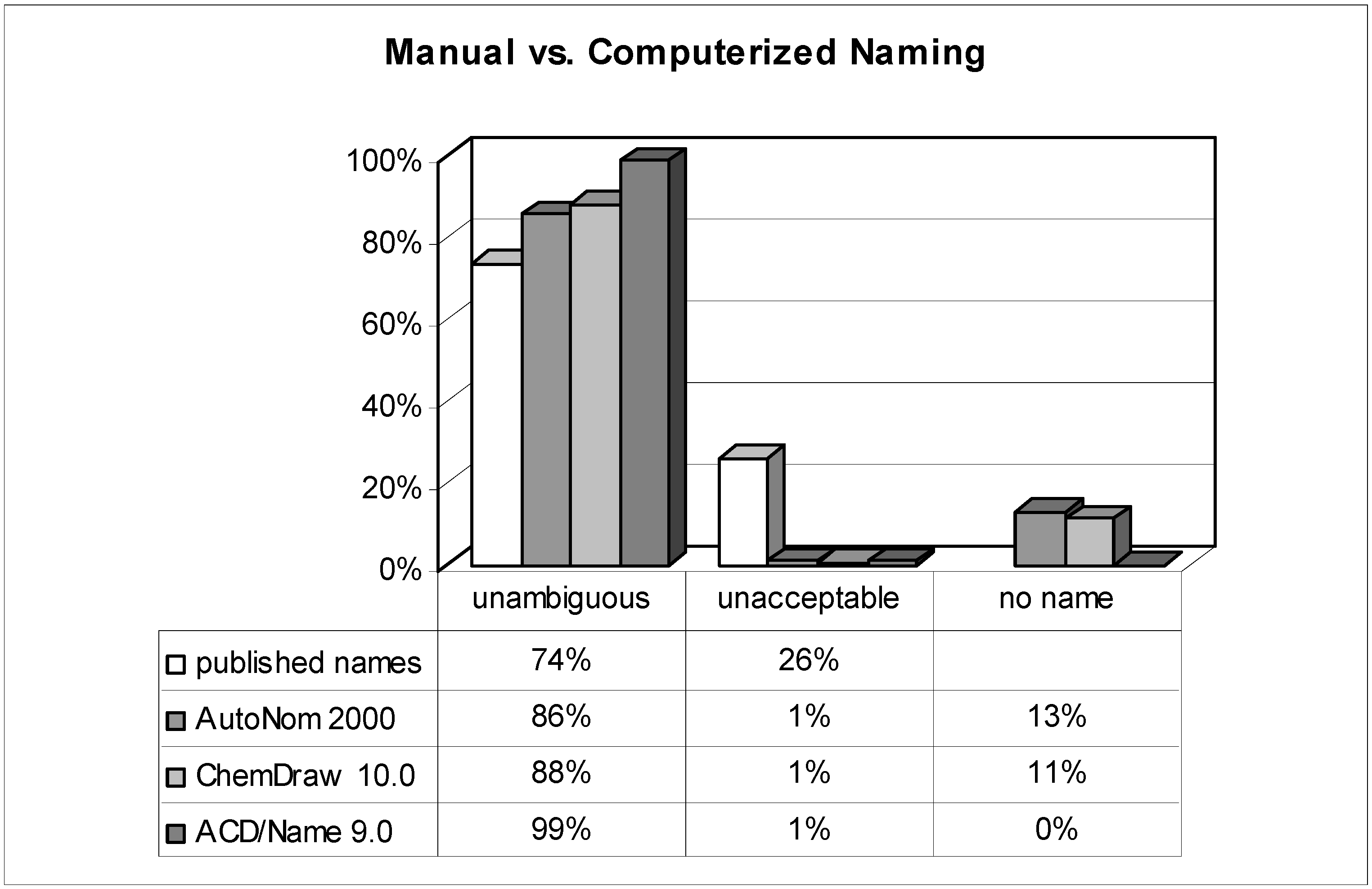
Comparison of published names with computer-generated ones (n = 303).
Results and Discussion
The analysis of more than 300 systematic names of organic compounds originally published by dozens of authors in different international journals provides very good insight into the authors' nomenclature skills when it comes to a real-life test. While the detailed results are shown in Figure 2 and Figure 3, the most surprising outcome was that roughly one quarter of all manually assigned names were deemed unacceptable and thus useless. This means that the substances were named incorrectly in such a way that is impossible to generate the described chemical structure solely by analyzing its proposed name! On the one hand, it seems to be tedious and unacceptable for any reader to waste his time with futile or even misleading names; on the other, it casts doubt on the reliability and thoroughness of other data provided. A frequently encountered weak point was related to stereochemistry.
Figure 3.
Quality of the unambiguous names.
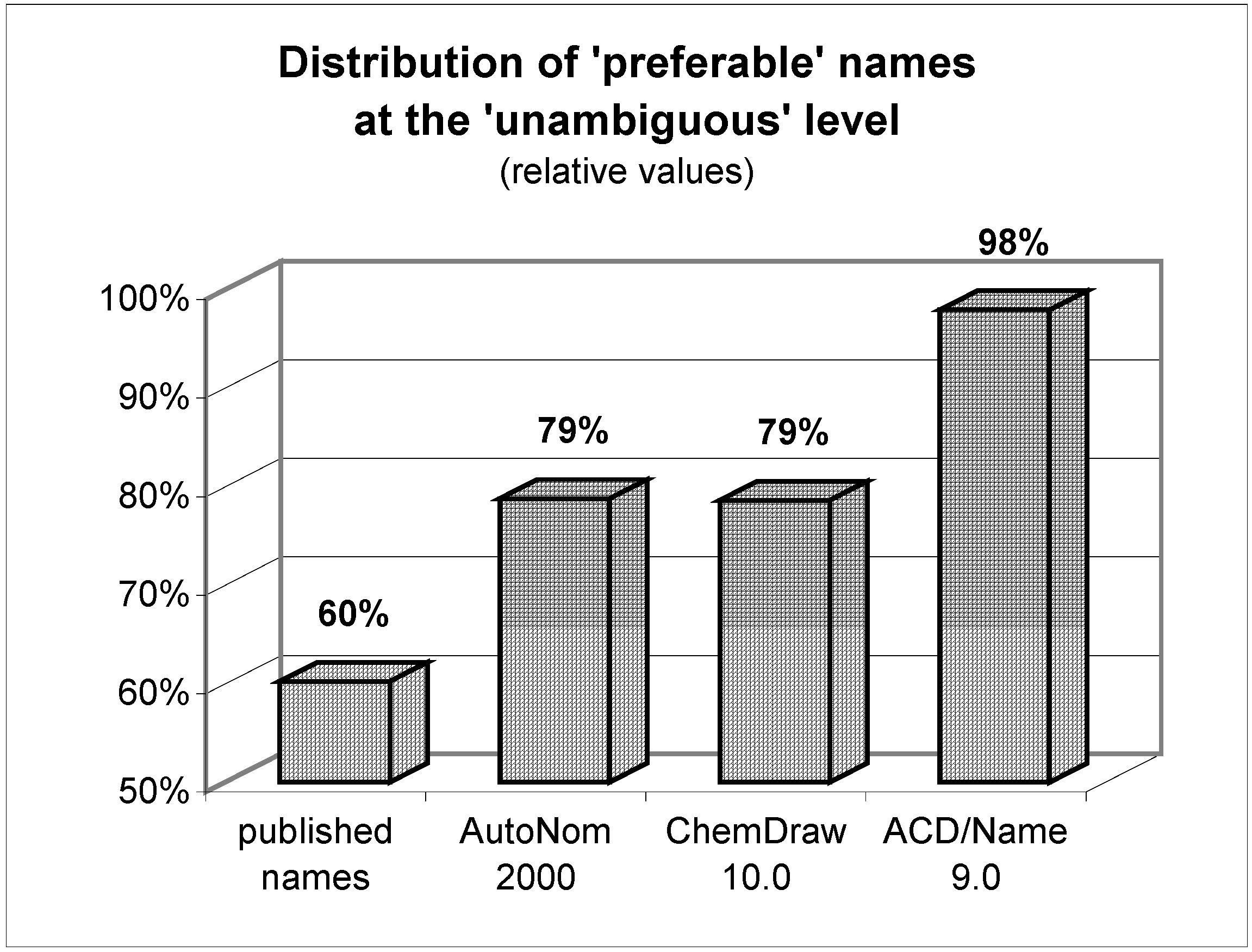
How shall we judge a chemist who, for example, claims to investigate the stereoselectivity of reactions if he does not even manage to name and communicate his molecules clearly to other researchers? After all, nearly half of all published names are regarded to be in full accordance with the systematic rules ('preferable').
While the reasons for the average scientists' apparently moderate or even poor knowledge in matters of systematic nomenclature rules remain unanswered, a possible way for improvement – prescription of extra lessons would not be realistic – is revealed by this investigation: nomenclature software. When reconsidering the results of the tested software programs and comparing them to the published names, the most striking advantage is that their failure to prevent 'unacceptable' names is quite rare (~1%). In a nutshell: as long as the nomenclature software generates a name, it's almost definitely an 'unambiguous' and hence interpretable one. Nevertheless, not every output name strictly follows the rules, and some differences in performance between the used nomenclature tools merit further discussion.
Although ChemDraw has developed its own nomenclature algorithm since version 8 (the previously implemented AutoNom 2000 was no longer updated and improved by MDL), both AutoNom and ChemDraw are still comparable in their performance. Their superiority to the naming skills of an average scientist is proven by the avoidance of irreproducible names. Even when the quality of the names is compared (the ratio of 'preferably' named compounds among the class of 'unambiguous') these software algorithms are superior to the humans' one, too. A negative point, particularly for ChemDraw, is the quite high rate (11%) of naming rejections. While ChemDraw refuses to name more challenging ring systems, AutoNom tries to solve these tasks by applying replacement or von Baeyer nomenclature operations which in most cases leads to needlessly tedious names. It is worth mentioning that in almost two thirds of the refused structures in AutoNom, the algorithm was unable to recognize the stereochemistry due to the undefined topology of a double bond. This restriction can be easily circumvented – redrawing the molecule with either a cis (Z) or trans (E) double bond and deleting the stereodescriptor from the name – and the 'no name' rate may then be lowered to 5%. In both packages the fundamental rules of Cahn, Ingold, and Prelog ('CIP-rules') [39] that determine the stereochemistry of a stereocenter are implemented and reliably handled. It can be summarized that both packages readily outperform the average scientist and both meet the requirements to be useful and reliable helpers for day-to-day name creation of simple and even of more advanced organics.
When the results of the other nomenclature software, ACD/Name (IUPAC), are examined an even more impressive performance of this software is revealed: no (nota bene!) structure's naming was refused; whereas the percentage of 'unacceptable' names was as low as with the other programs (1%). Furthermore, the quality of names that are generated from ACD/Name is second to none; although possible, it was really difficult to find generated names that did not deserve being called 'preferable' (97%). On the one hand, the basic nomenclature operations are almost completely supported; on the other, the programmers did dare to tackle some of the more sophisticated areas of nomenclature. Extensive implementation of the rules for the nomenclature of complicated polycyclic ring systems, amino acids, and carbohydrates significantly widens the range of structural elements that can be named satisfactorily. Other very specialized compounds like coordination compounds (typically in inorganic chemistry), polymers, and many classes of natural products (alkaloids, steroids, etc.) are also supported to a considerable extent.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Agreement of generated Index names (ACD/Index Name 9.0) with the officially registered ones (CAS).
| ACD/Index Name 9.0 | ||
| names tested | 303 | 100% |
| names generated | 301 | 99% |
| no name generated | 2 | 1% |
| identical to CAS name | 266 | 88% |
| different to CAS name | 35 | 12% |
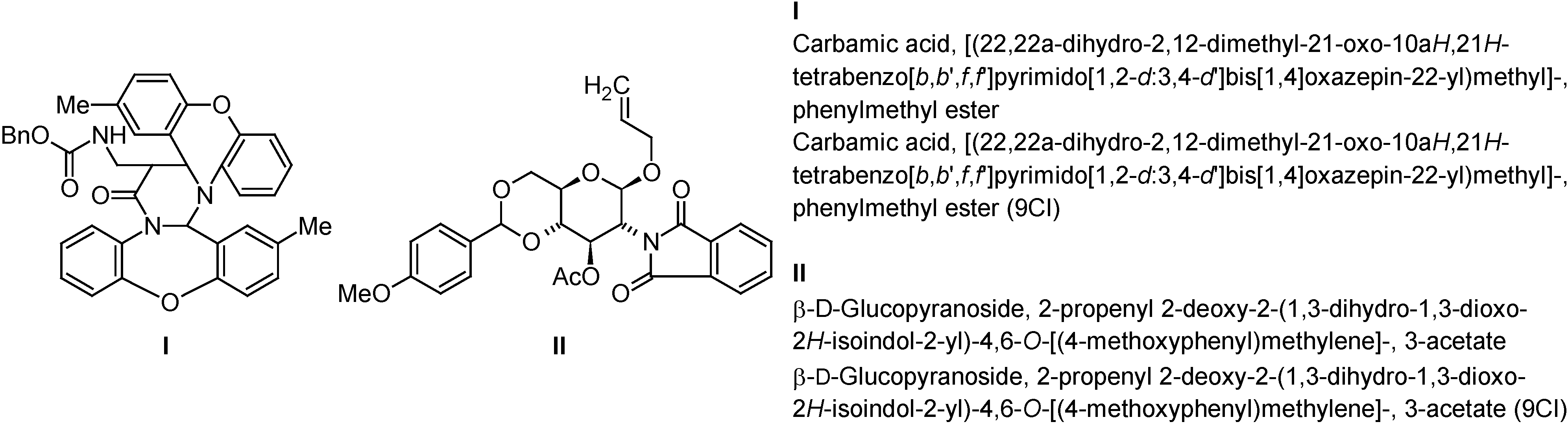
The results of Index name generation with ACD/Name according to CAS rules are shown in Table 1. Since the CAS rules allow only a single name for every structure the primary goal of the software must lie on the generation of an identical name, which was achieved 88% of the time. Two impressive examples of identically named structures are given in Figure 4.
Figure 4.
Two impressive examples of generated Index names (ACD/Name) that are identical to the officially registered ones (CAS; CI ... collective index).
Figure 4.
Two impressive examples of generated Index names (ACD/Name) that are identical to the officially registered ones (CAS; CI ... collective index).
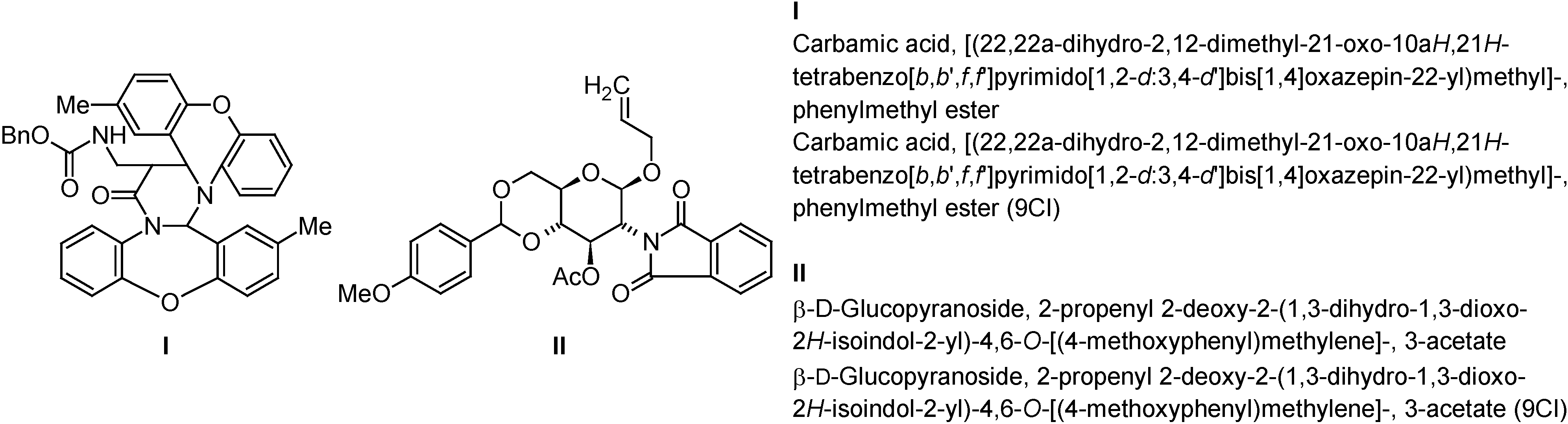
In about half of the other cases, an alternative, but only slightly different name was generated; e.g. the unessential locant for the stereodescriptor in #61 (Figure 1):
- Phosphonic acid, [(2-chlorophenyl)[[(1E)-3,3-dicyano-2,2-dimethylpropylidene]amino]methyl]-, diethyl ester (ACD/Name) versus
- Phosphonic acid, [(2-chlorophenyl)[(E)-(3,3-dicyano-2,2-dimethylpropylidene)amino]methyl]-, diethyl ester (CAS, 9CI).
Conclusions
This paper investigated the quality of published chemical names and demonstrated that they suffer from ambiguity and low quality in a considerable number of cases. Although clear rules for name generation exist, they are obviously not followed by chemists. An easy method to improve this unsatisfactory situation is the use of modern nomenclature software, because all three tested programs were far better than the average chemist. It remains to be seen whether these results will enhance the behavior of editors and authors in systematic naming.
Although preferable, it is often impossible to use (commercial) nomenclature services provided by experts. Nevertheless, a practical means for improvement could be the combination of manual naming and computer cross-checking, by entering the proposed name into a structure generation tool like 'Name=Struct' [40] (integrated into ChemDraw) or 'Name to Structure' (part of ACD/Name). What makes these tools really helpful is their tolerance of different nomenclature styles and even some errors and typos. If the generated structure agrees with the original one, the molecule can be named with the nomenclature tool and the computerized name can be compared with the manual one. Through the use of this procedure typos become apparent and misleading or wrong names can be filtered out and replaced. This method should become a routine assisting tool during the preparation/publication of (high-quality) scientific articles containing chemical names (journals, patents, etc.).
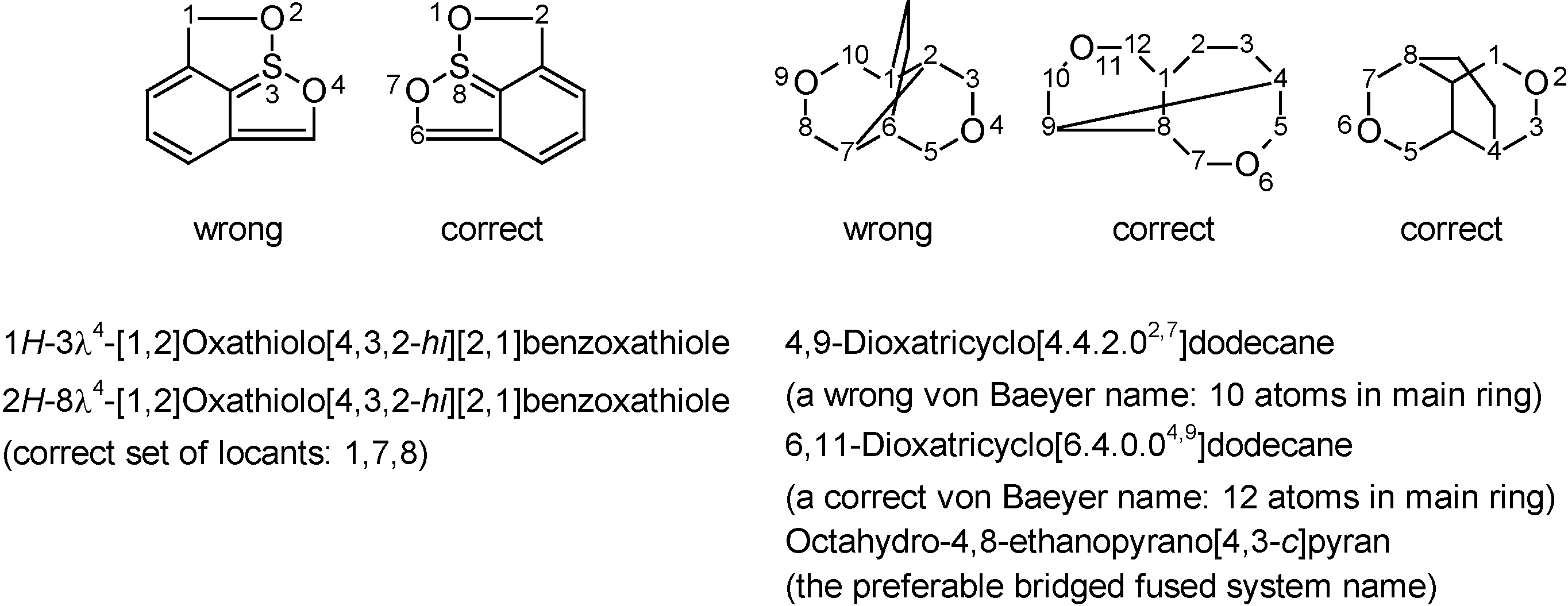
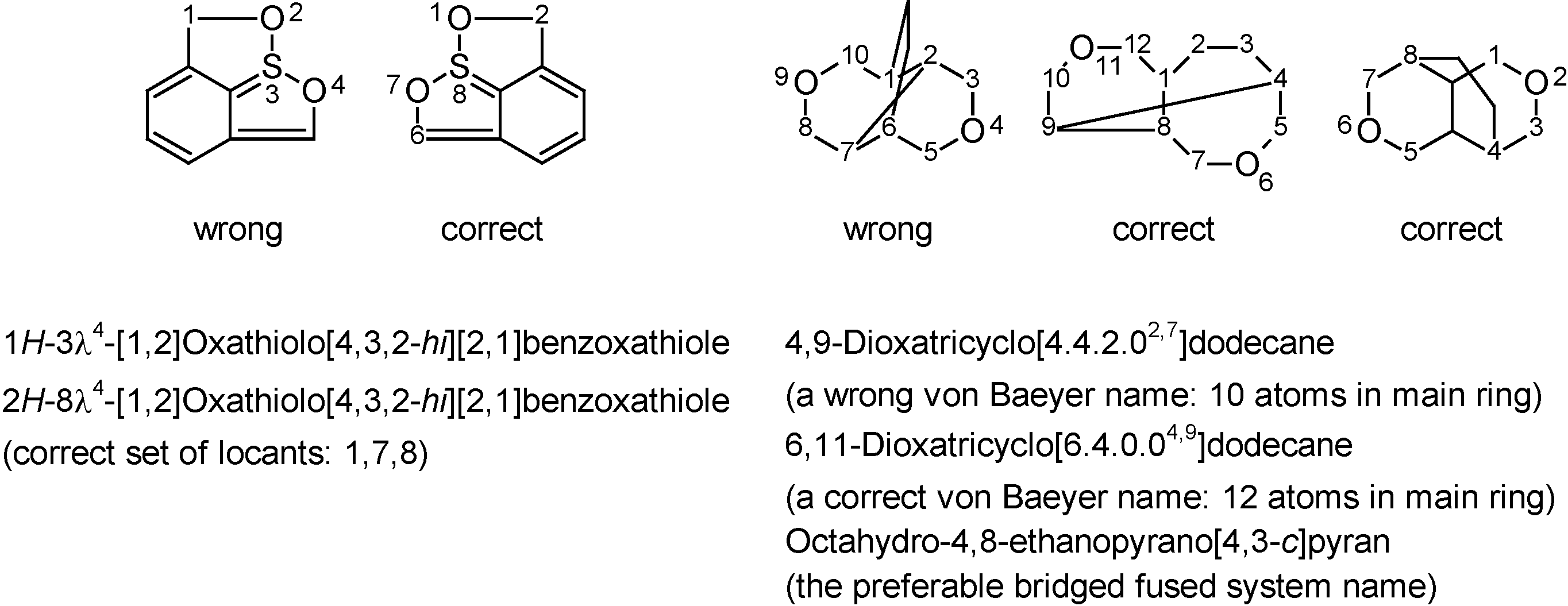
Another advantage of computerized nomenclature algorithms is their impartiality towards the graphical representation of a structure. While humans are easily mislead by a specific view of a 2D or 3D structure, computers 'ignore' this kind of subjectivity. The two examples of Figure 5 – extracted (and simplified) from a provisional IUPAC recommendation in which they were originally incorrectly named – are typical examples of structures that humans (average scientists as well as nomenclature experts) might attempt to name differently depending on representation considered. The software generates only a single name, regardless of how the structure was drawn. In spite of the many advantages that state-of-the-art nomenclature software offers for every-day molecules, it is necessary to stress its limitations. As with every computer program: algorithms that have not been programmed will not be available. Hence, software – at least for now – cannot be applied to complicated areas of nomenclature that are very specific or quite uncommon, e.g. very complicated bridged fused systems [41] or rotaxanes [11]. Although errors or examples of bad programming are rare and will typically be corrected as a matter of course in subsequent releases, there is currently no way around contacting nomenclature professionals when (such) compounds have to be named with absolute certainty (e.g. in patents).
Figure 5.
Originally incorrectly named examples extracted from a provisional nomenclature recommendation [8] that demonstrate the impartiality of computer software towards structure representation, whereas humans may attempt to name the different representations differently (correct names were generated with ACD/Name).
Figure 5.
Originally incorrectly named examples extracted from a provisional nomenclature recommendation [8] that demonstrate the impartiality of computer software towards structure representation, whereas humans may attempt to name the different representations differently (correct names were generated with ACD/Name).
To help the readers in determining if and which software fits best for specific needs, a comparison of the programs utilized can be found in Table 2, which ranks the supported functional classes in detail. This evaluation is based upon the author's own experience during this thorough testing which included the naming of examples from the corresponding IUPAC recommendations. Since quantification in these matters is always difficult, and since subjective impressions cannot be excluded, this ranking should be regarded only as a general reference. The individual testing of these programs is recommended. Since free or trial versions can be downloaded from the companies' websites [18,19,20], expense should not be an excuse for using bad names in scientific literature. Besides, the costs for the commercial packages, while sometimes seeming to be quite high at the first glance, are minor compared to other analytical equipment used for proper compound characterization (NMR, HPLC, MS, etc.). Moreover, nomenclature software tools seem to be suitable for educational purposes, since they greatly assist the learning and understanding of systematic nomenclature.
| AutoNom 2000 | ChemDraw 10.0 | ACD/Name 9.0 | |
|---|---|---|---|
| functional groups | + + + | + + + + + | + + + + + |
| stereochemistry | + + + + | + + + + + | + + + + + |
| hydrocarbon chains | + + + + + | + + + + + | + + + + + |
| heteroatom chains | −a | + + + + | + + + + + |
| multiplicative nomenclature | −a | + + + + | + + + |
| monocycles | + + + + + | + + + + | + + + + + |
| fused polycycles | + + + | + + + | + + + + + |
| von Baeyer polycyclesb | + + + | + + | + + + + + |
| bridged fused systems | −a | −a | + + + |
| spirocycles | + + + | + + + | + + + + + |
| ring assembliesc | + + + + + | + + + | + + + + + |
| 'indicated' hydrogen | + + + + | + + + | + + + + + |
| non-standard valencesd | + + + | −a | + + + + + |
| salts & radicals & ions | + + | + + + | + + + + + |
| biochemicals & natural productse | −a | −a | + + + |
| organometallicsf | −a | + | + + + |
| polymers | −a | −a | + + + |
| structure generation toolg | −a | + + + + + | + + + + |
| a not supported; b von Baeyer polycycles are bridged non-fused systems, e.g. bicyclo[3.2.1]octane; c e.g. 2,2':6',4''-terpyridine; d e.g. thiophene 1-oxide or 1λ5-phosphinane; e ACD/Name 9.0 supports the following classes on a more or less limited scope: carbohydrates, amino acids, steroids, alkaloids, terpenoids; f ACD/Name 9.0 supports the naming of various coordination compounds with neutral and anionic ligands. Coordination sites are specified according to κ- and η-conventions; g these tools generate chemical structures from (semi)systematic/trivial names. | |||
Acknowledgements
The author is grateful to the referees of a previous version of the manuscript for some useful comments as well as to the IUPAC Division VIII (Chemical Nomenclature and Structure Representation Division) and its president, Dr. G. P. Moss, for discussions. Moreover, the author thanks Cambridge Soft, ACD/Labs, and Scienceserve [42] for providing actual versions of their software packages and for their technical support.
References and Notes
- Williams, A.; Yerin, A. The need for systematic naming software tools for exchange of chemical information. Molecules 1999, 4, 255–263. [Google Scholar] [CrossRef]
- Hellwinkel, D. Systematic Nomenclature of Organic Chemistry: A Directory to Comprehension and Application of its Basic Principles; Springer: Berlin, 2001. [Google Scholar]
- Hellwich, K.-H. Chemische Nomenklatur; Govi: Eschborn, 2002. [Google Scholar]
- Fennell, R.W. History of IUPAC 1919–1987; Blackwell Science: Oxford, 1994. [Google Scholar]
- International Union of Pure and Applied Chemistry. Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H, 1979 Edition; Pergamon: Oxford, 1979. [Google Scholar]
- International Union of Pure and Applied Chemistry (IUPAC); Organic Chemistry Division, Commision on Nomenclature of Organic Chemistry (III.1). A Guide to IUPAC Nomenclature of Organic Compounds: Recommendations 1993; Panico, R., Powell, W.H., Richer, J.C., Eds.; Blackwell Science: Oxford, 1993; [Chem. Abstr. 1994, 120, 297652]. [Google Scholar]
- Favre, H.A.; Hellwich, K.-H.; Moss, G.P.; Powell, W.H.; Traynham, J.G. Corrections to a guide to IUPAC nomenclature of organic compounds (IUPAC recommendations 1993). Pure Appl. Chem. 1999, 71, 1327–1330. [Google Scholar] [CrossRef]
- Favre, H.A.; Powell, W.H. Nomenclature of Organic Chemistry [Provisional Recommendations, IUPAC]. Available online: http://www.iupac.org/reports/provisional/abstract04/favre_310305.html (accessed on October 2006).
- Favre, H.A.; Hellwinkel, D.; Powell, W.H.; Smith, H.A., Jr.; Tsay, S.S.-C. Phane nomenclature. Part II. Modification of the degree of hydrogenation and substitution derivatives of phane parent hydrides (IUPAC recommendations 2002). Pure Appl. Chem. 2002, 74, 809–834. [Google Scholar]
- Cozzi, F.; Powell, W.H.; Thilgen, C. Numbering of fullerenes. Pure Appl. Chem. 2005, 77, 843–923. [Google Scholar] [CrossRef]
- Yerin, A.; Metanomski, W.V.; Moss, G.P.; Wilks, E.S.; Harada, A. Nomenclature for Rotaxanes. Submitted to Pure Appl. Chem. [Provisional Recommendations, IUPAC]. Available online: http://ibiblio.lsu.edu/main/iupac/reports/provisional/abstract05/yerin_300406.html (accessed on October 2006).
- Kirby, G.H.; Polton, D.J. Systematic chemical nomenclatures in the computer age. J. Chem. Inf. Comput. Sci. 1993, 33, 560–563. [Google Scholar] [CrossRef]
- For example: The Journal of Organic Chemistry, Guidelines for Authors 2006; (Anon. J. Org. Chem. 2006, 71, 1A–10A).
- Garfield, E. Chemico-linguistics: computer translation of chemical nomenclature. Nature 1961, 192, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.L. AUTONOM: system for computer translation of structural diagrams into IUPAC-compatible names. 1. General design. J. Chem. Inf. Comput. Sci. 1990, 30, 324–32. [Google Scholar] [CrossRef]
- Goebels, L.; Lawson, A.J.; Wisniewski, J.L. AUTONOM: system for computer translation of structural diagrams into IUPAC-compatible names. 2. Nomenclature of chains and rings. J. Chem. Inf. Comput. Sci. 1991, 31, 216–25. [Google Scholar] [CrossRef]
- Wisniewski, J.L. Digital naming of organic compounds: on some successful algorithms. Hypernews 1997, 2, 22–29. [Google Scholar]
- ISIS Draw 2.5 with AutoNom 2000. MDL Information Systems, Inc.: San Leandro CA, USA, www.mdl.com; 2002.
- ChemDraw Ultra 10.0. CambridgeSoft Corporation: Cambridge MA, USA, www.cambridgesoft.com; 2005.
- ACD/Name, version 9.08. Advanced Chemistry Development, Inc.: Toronto ON, Canada, www.acdlabs.com; 2006.
- IUPAC NameIt. Bio-Rad Laboratories: Philadelphia PA, USA, www.bio-rad.com.
- Lexichem, version 1.5. OpenEye Scientific Software, Inc.: Santa Fe NM, USA, www.eyesopen.com; 2006.
- Nomenclator. ChemInnovation Software, Inc.: San Diego, CA, USA, www.cheminnovation.com; 2006.
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J. Chem. Inf. Comput. Sci. 2004, 44, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Engel, T. Valenzen, Bindungen und Orbitale – Struktureditoren. Nachr. Chem. 2003, 51, 450–453. [Google Scholar] [CrossRef]
- Zielesny, A. Chemistry Software Package ChemOffice Ultra 2005. J. Chem. Inf. Model. 2005, 45, 1474–1477. [Google Scholar] [CrossRef]
- Irwin, J.J. Software Review: ChemOffice 2005 Pro by CambridgeSoft. J. Chem. Inf. Model. 2005, 45, 1469. [Google Scholar]
- Mendelsohn, L.D. ChemDraw 8 Ultra, Windows and Macintosh Versions. J. Chem. Inf. Comput. Sci. 2004, 44, 2225–2226. [Google Scholar] [CrossRef]
- Selliah, R.D. IUPAC Name Pro 4.5 with Name to Structure Module. J. Am. Chem. Soc. 2001, 123, 6463. [Google Scholar]
- Koch, R.; Lemmler, M. Nomenklatur in der Chemie: The next generation. Nachr. Chem. 2000, 48, 642–646. [Google Scholar] [CrossRef]
- Vogt, J. Chemische Nomenklatur per Mausklick. Nachr. Chem. 2005, 53, 428–431. [Google Scholar] [CrossRef]
- For the latter two journals the 2005 issues had not been fully indexed by the CAS when this work was started.
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- Maldonado, A.G.; Doucet, J.P.; Petitjean, M.; Fan, B.-T. Molecular similarity and diversity in chemoinformatics: From theory to applications. Mol. Diversity 2006, 10, 39–79. [Google Scholar] [CrossRef]
- 'Struct=Name' is a new proprietary algorithm that replaces the AutoNom one used in earlier versions of ChemDraw.
- 'Use Retained Replacements', 'Advanced Enclosing Marks', No 'Forward Locants Position', Hantzsch–Widman New Stems', 'Extended Fused List', No 'New Functional Groups Names', 'Stereoconfiguration: Absolute', no 'Stereo Wedge Direction', no 'Name Preferred Tautomeric Form', 'Refuse To Name: Fused Multiparent Systems, Complex Bridged Fused Systems', Complex Multiplicative Structures', 'Use Biochemical Names: Steroids, Alkaloids, Terpenes, Carbohydrates, Amino Acids, Peptides', 'Select Language: English'. For both nomenclature tools 'Capitalization First Letter of Name'' was chosen and for ACD/Name Index [31] 'Name Preferred Tautomeric Form' was enabled.
- ACD/Labs has developed its naming algorithms alone and calls the CAS-like names 'Index names'.
- Wagner, B.A. SciFinder Scholar 2006: An Empirical Analysis of Research Topic Query Processing. J. Chem. Inf. Model. 2006, 46, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. Spezifikation der molekularen Chiralität. Angew. Chem. Int. Ed. 1966, 5, 385–415. [Google Scholar]
- Brecher, J. Name=Struct: A Practical Approach to the Sorry State of Real-Life Chemical Nomenclature. J. Chem. Inf. Comput. Sci. 1999, 39, 943–950. [Google Scholar] [CrossRef]
- Moss, G.P. Nomenclature of fused and bridged fused ring systems. Pure Appl. Chem. 1998, 70, 43–216. [Google Scholar] [CrossRef] [Green Version]
- ScienceServe GmbH, Pegnitz, Germany (www.scienceserve.com). ScienceServe is the local ACD distributor for Germany, Austria, and Switzerland.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Eller, G.A. Improving the Quality of Published Chemical Names with Nomenclature Software. Molecules 2006, 11, 915-928. https://doi.org/10.3390/11110915
AMA Style
Eller GA. Improving the Quality of Published Chemical Names with Nomenclature Software. Molecules. 2006; 11(11):915-928. https://doi.org/10.3390/11110915
Chicago/Turabian StyleEller, Gernot A. 2006. "Improving the Quality of Published Chemical Names with Nomenclature Software" Molecules 11, no. 11: 915-928. https://doi.org/10.3390/11110915