Synthesis and Anticancer Activities of Novel 1,4-Disubstituted Phthalazines

School of Pharmaceutical Engineering, Shenyang Pharmaceutical University, 103 Wenhua Road,
Shenhe District, 110016 Shenyang, Liaoning, P. R. China
*
Author to whom correspondence should be addressed.
Molecules 2006, 11(7), 574-582; https://doi.org/10.3390/11070574
Submission received: 25 June 2006
/
Revised: 12 July 2006
/
Accepted: 13 July 2006
/
Published: 27 July 2006
Abstract
:A series of novel 1-anilino-4-(arylsulfanylmethyl)phthalazines were designed and synthesized. The structures of all the compounds were confirmed by IR, 1H-NMR, elemental analysis and MS. The analogues 1-(3-chloro-4-fluoroanilino)-4-(3,4-difluorophenylthio-methyl)phthalazine (12) and 1-(4-fluoro-3-trifluoromethylanilino)-4-(3,4-difluorophenyl-thiomethyl)phthalazine (13) showed higher activity than a cisplatin control when tested in vitro against two different cancer cell lines using the microculture tetrazolium method (MTT) method.
Introduction
Phthalazine derivatives, like the other members of the isomeric benzodiazine series, have been widely applied as therapeutic agents due to their anticonvulsant, cardiotonic, vasorelaxant and anti-inflammatory properties [1,2]. To our knowledge, however, there have been no reports on the anticancer activities of 1-anilino-4-arylsulfanylmethylphthalazines. We describe here the synthesis of some novel 1-anilino-4-arylsulfanylphthalazine derivatives 9-19, several of which exhibited higher activity than the cisplatin control.
Phthalazines were previously synthesized from 2-aryl-3-hydroxyinden-1-ones or β-diketones by condensation with hydrazine hydrate [2,3,4,5]. However, these routes do not allow for the desired incorporation of thiophenylmethyl groups into the phthalazine 4-position. This report describes a convenient access to 1,4-disubstituted phthalazines with such substituents. The compounds obtained in were characterized by IR, 1H-NMR, MS and elemental analysis and their anticancer activities were evaluated in vitro.
Results and Discussion
Chemistry
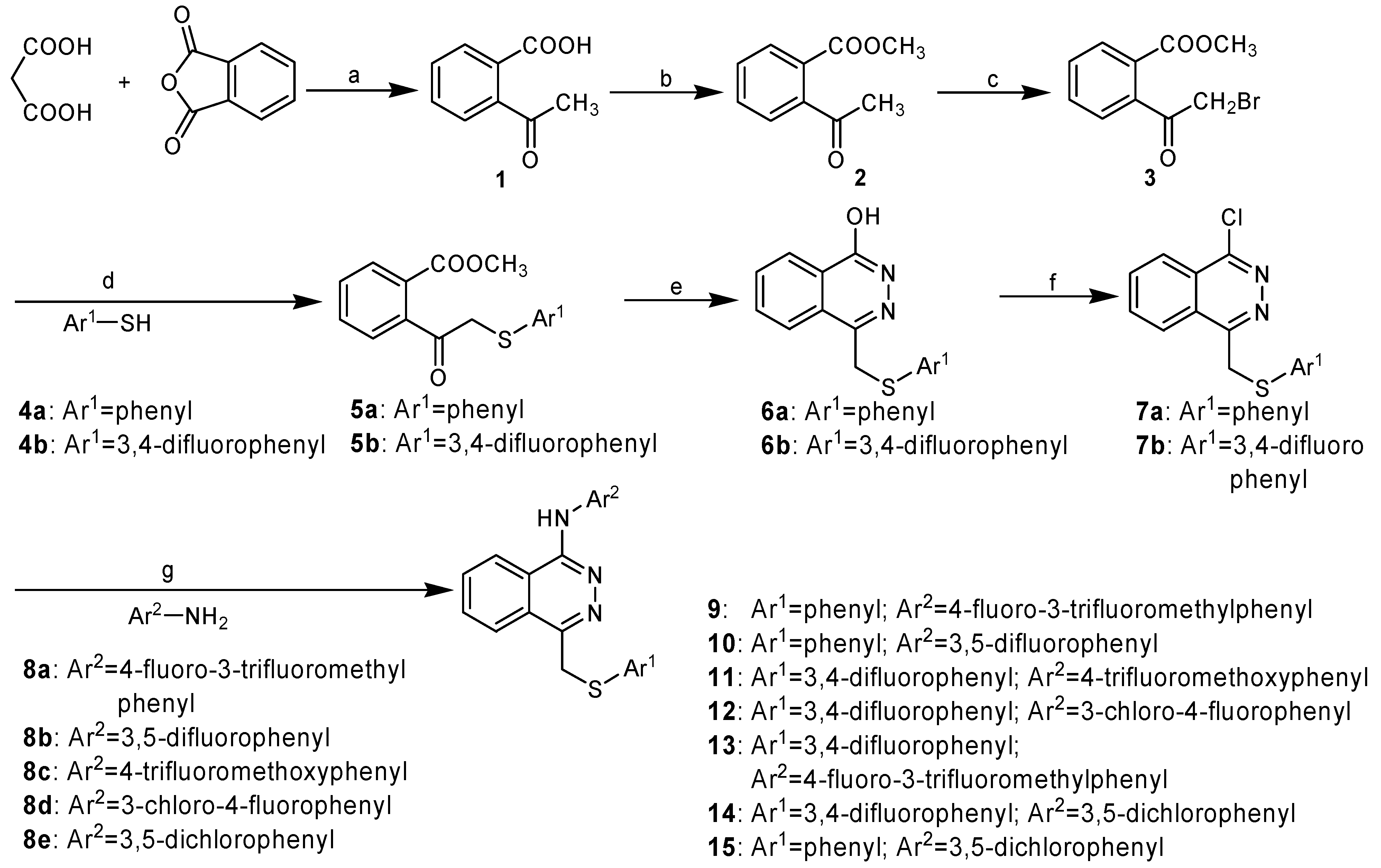
Our syntheses of the requisite phthalazines are illustrated in Scheme 1 and Scheme 2. Refluxing phthalic anhydride and malonic acid in pyridine gave 2-acetylbenzoic acid (1) [6], which was then esterified with dimethyl sulfate. In the next step, the acetyl group was brominated with phenyltrimethyl-ammonium tribromide (PTT), a selective brominating reagent for ketones or ketals [7], to give intermediate 3, which facilitated the introduction of various thiophenol substituents. It should be noted that side-products could be formed if bromine was used as the halogen source. Compound 3 was then treated with thiophenol or 3,4-difluorothiophenol using K2CO3 as base. Cyclization of 5a/5b with hydrazine hydrate led to the generation in 90-96% yields of the phthalazines 6a/6b, which were treated with POCl3 to give 1-chloro-4-substituted-phthalazines 7a and 7b. Treatment of 7a/7b with substituted anilines provided the target compounds 9-15.
Scheme 1.
Synthesis of compounds 9-15.

Reagents and conditions: a) Py, reflux, 3 h; b) Me2SO4, K2CO3, acetone, reflux, 3 h; c) PTT, THF, r.t., 15 h; d) K2CO3, CH3OH, r.t, 1.5 h; e) H2NNH2.H2O, CH3OH, reflux, 5 h; f) POCl3, 110 oC, 3 h; g) i-PrOH, 50 oC, 3 h
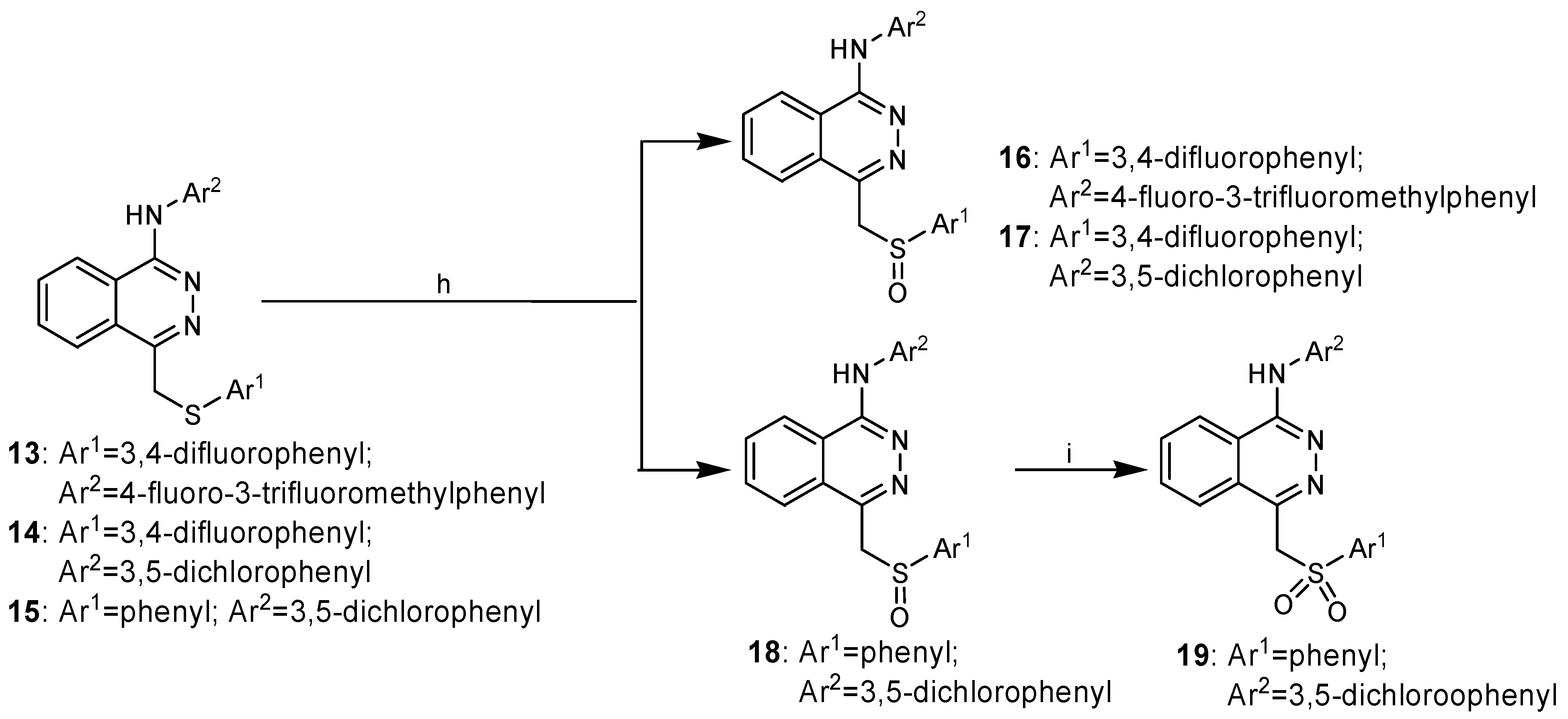
The target compounds 16-18 could be obtained by oxidization of the corresponding compounds 13-15 with H2O2. However, the sulfanyl substituted phthalazine derivatives could not be easily converted to the corresponding sulfonyl ones. MCPBA has been used as the oxidizing reagent in some cases [8], but in our synthetic route this could easily lead to an undesired N-oxidized side-product, due to the presence of the amino groups. We have found, however, that H2O2/Na2WO4 was very effective for oxidizing sulfanyl to sulfonyl groups in good yield and high purity.
Scheme 2.
Synthesis of compounds 16-19.
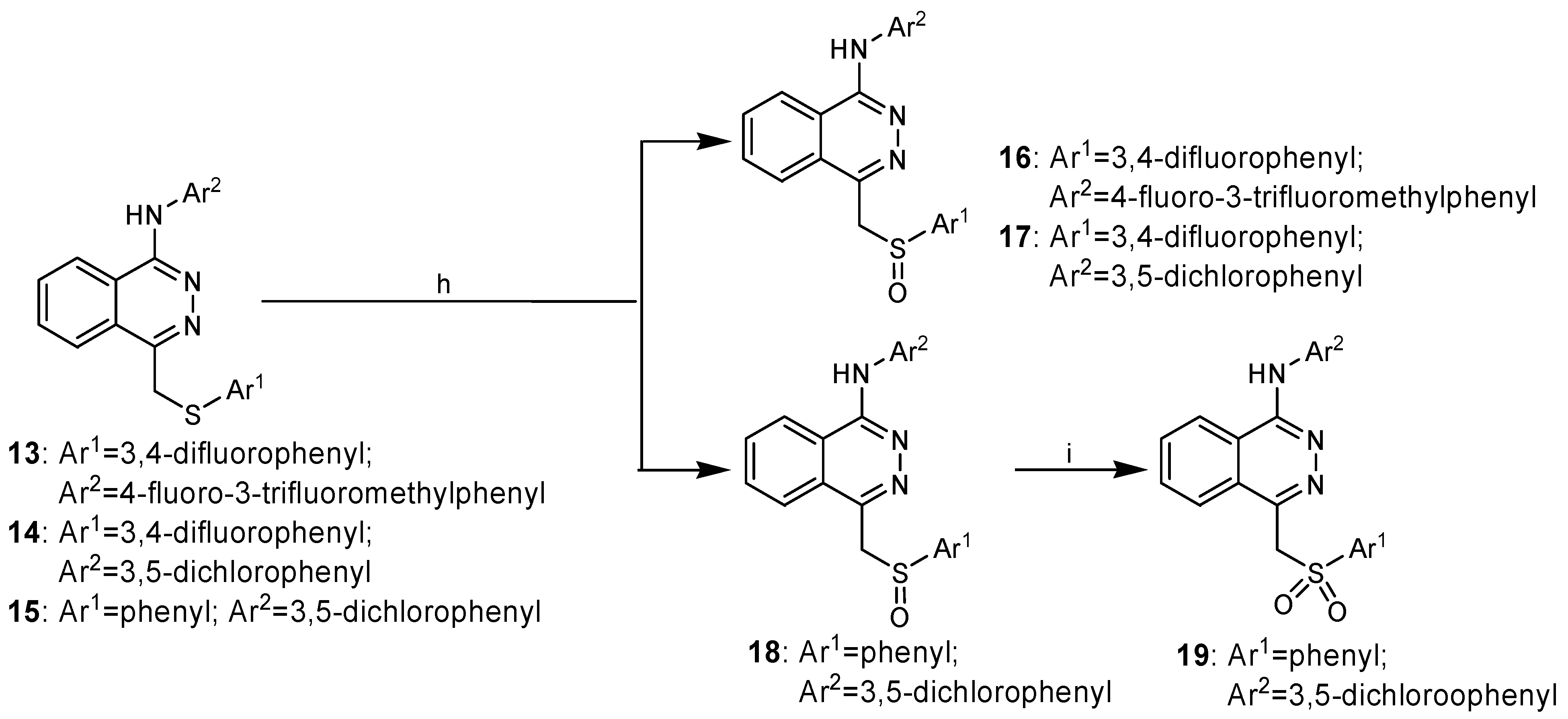
Reagents and conditions: h) H2O2, HOAc, r.t., 16 h; i) Na2WO4·2H2O, H2O2, CH3OH, r.t., 18 h
Anticancer activities
The anticancer activities of compounds 9-19 were evaluated in vitro by the MTT method and the results are summarized in Table 1.

{kind=link}
{kind=link}
| Compd. | IC50 (μM) | Compd. | IC50 (μM) | ||
|---|---|---|---|---|---|
| Bel-7402 | HT-1080 | Bel-7402 | HT-1080 | ||
| 9 | 146.8 | 83.9 | 15 | 91.3 | 126.5 |
| 10 | 116.1 | 163.4 | 16 | 164.2 | 122.4 |
| 11 | 56.2 | 38.9 | 17 | 125.0 | 133.6 |
| 12 | 32.4 | 25.4 | 18 | 198.6 | 158.4 |
| 13 | 30.1 | 25.8 | 19 | 244.0 | 169.9 |
| 14 | 69.2 | 60.3 | cisplatin | 73.3 | 63.3 |
Compounds 12 and 13 showed more in vitro activity than cisplatin against the two cancer cell lines tested. The phthalazine derivative 11 showed activity comparable to cisplatin. The remaining compounds exhibited slight to moderate activities.
Conclusions
A series of novel 1,4-disubstituted phthalazines have been prepared. From the biological test results the following conclusions can be reached about their structure-activity relationships: (a) incorporation of a substituted thiophenol group into position 4 of the phthalazine ring appears to increase anticancer activity, compared to that of an unsubstituted thiophenol; (b) replacement of a sulfanyl with sulfinyl or sulfonyl groups decreases the anticancer activity. Further investigations are in process.
Experimental
General
Melting points were determined by the capillary tube method, and the thermometer was uncorrected. Mass spectra were obtained on an Agilent 1100 HPLC-MS instrument. 1H-NMR spectra were run in DMSO-d6, with TMS at the internal standard, on a Bruker ARX-300 instrument operating at 300 MHz. IR spectra (KBr disks) were recorded on a Bruker IFS 55 instrument. Elemental analysis was performed with a Carlo-Erba 1106 Elemental analysis instrument.
Chemistry
2-Acetylbenzoic acid (1)
A mixture of phthalic anhydride (22.2 g, 0.15 mol), malonic acid (18.7 g, 0.18 mol) and pyridine (17.3 mL, 0.18 mol) was refluxed for 3 h. The resulting mixture was then cooled to 30 oC, water (160 mL) was added and the mixture was stirred for 30 min. The insoluble material was filtered off and the filtrate was treated with concentrated HCl to pH 3-4. Filtration and recrystallization from chloroform gave 16.8 g (68%) of 1, m.p. 113-114 oC (lit. [6] 114-115 oC); MS: m/z 165 (MH+); IR (cm-1): 3450.1 (OH), 1685.2, 1676.3 (C=O), 1639.7, 1590.6 (C=C).
Methyl 2-acetylbenzoate (2)
A mixture of 2-acetylbenzoic acid (1, 100.0 g, 0.61 mol), dimethyl sulfate (92.0 g, 0.73 mol) and K2CO3 (50.0 g, 0.37 mol) was refluxed for 3 h in acetone. The reaction mixture was cooled to room temperature and filtered. The filtrate was distillated under reduced pressure collecting the fraction with b.p. 86-92 oC/6 mmHg, which gave product 2 as a light yellow oil, 84 g (77%); GC purity: 97.6%; MS: m/z 179 (MH+).
Methyl 2-(bromocarbonyl)benzoate (3)
To a solution of 2 (100.0 g, 0.56 mol) in anhydrous tetrahydrofuran (200 mL), a solution of PTT (210.5 g, 0.56 mol) in anhydrous tetrahydrofuran (80 mL) was added dropwise. During this addition a white precipitate was formed and the solution became yellow. After stirring at room temperature for 15 h, the resulting mixture was filtered. The filtrate was stirred into a mixture of petroleum ether/water (200 mL, 1:1 v/v), then separated and concentrated to give 116.4 g (80%) of 3; m.p.132-133 oC; MS: m/z 257, 259 (MH+); IR (cm-1): 2938.3 (CH), 1689.2, 1681.3 (C=O), 1647.7, 1602.6 (C=C); Anal. Calcd. for C10H9BrO3: C 46.72, H 3.53, Br 31.08; Found: C 46.60, H 3.34, Br 31.19.
Methyl 2-(2-(phenylthio)acetyl)benzoate (5a)
To a mixture of K2CO3 (8.3 g, 0.06 mol) and 4a (11 g, 0.1 mol) in methanol (150 mL), a solution of 3 (25.7 g, 0.1 mol) in acetone (200 mL) was added dropwise while the temperature was kept below 0 oC. The reaction mixture was stirred for an additional 1.5 h at this temperature. After filtration and concentration, the residue was dissolved in dichloromethane (200 mL). The organic phase was washed with saturated sodium carbonate solution (100 mL×3) and dried with MgSO4. Concentration gave 5a as an oil, 23.5 g (82%, GC purity: 96.9%), which could be used in next step without purification.
Methyl 2-(2-(3,4-difluorophenylthio)acetyl)benzoate (5b)
Prepared using 4b as described for 5a; yellow oil, 27.5 g, (86%), GC purity: 98.3%.
1-Hydroxy-4-phenylthiomethylphthalazine (6a)
Hydrazine hydrate (15.6 g, 80%, 0.25 mol) was added into a solution of 5a (24.0 g, 84 mmol) in methanol (100 mL). The mixture was refluxed for 5 h. After cooling to room temperature, a solid mass precipitated. Filtration and recrystallization from ethyl acetate gave 6a, 20.4 g, (91%), m.p. 146-148 oC; MS: m/z 269 (MH+); IR (cm-1): 3610.3 (OH), 1611.1, 1549.5 (C=C); 1H-NMR: δ 4.89 (s, 2H, CH2), 7.27-7.31 (m, 3H, Ph-3H), 7.42 (d, J=7.0 Hz, 2H, Ph-2H), 8.17-8.25 (m, 2H, phthalazinyl-2H), 8.42 (d, J=8.0 Hz, 1H, phthalazinyl-H), 8.79 (d, J=8.0 Hz, 1H, phthalazinyl-H).
1-Hydroxy-4-(3,4-difluorophenyl)thiomethylphthalazine (6b)
Prepared using 5b as described for 6a, 24.2 g, (95%), m.p. 163-164 oC; MS: m/z 305 (MH+); IR (cm-1): 3408.3 (OH), 1613.1, 1567.3, 1509.6 (C=C); 1H-NMR: δ 4.85 (s, 2H, CH2), 7.23 (b, 1H, Ph-H), 7.59-7.67 (m, 2H, Ph-2H), 8.20-8.27 (m, 2H, phthalazinyl-2H), 8.47-8.49 (d, J=8.1 Hz, 1H, phthalazinyl-H), 9.04-9.05 (d, d, J=8.1 Hz, 1H, phthalazinyl-H).
1-Chloro-4-phenylthiomethylphthalazine (7a)
Phosphorus oxychloride (36.8 g, 0.24 mol) was added dropwise into a solution of 6a (104.5 g, 0.39 mol) in pyridine (37 mL, 0.47 mol). The mixture was slowly heated to 110 oC and stirred for 1h. After cooling to 50 oC, chloroform (100 mL) and cold water (100 mL) were added. The biphasic mixture was stirred for 30 min and the layers were separated. The organic layer was washed with 5% sodium bicarbonate solution, dried and concentrated. The residue was triturated with diethyl ether by stirring for 3 h to give a suspension that was filtered to afford 7a, 92.8 g, (83%), m.p. 151-153 oC; MS: m/z 285, 287 (MH+); IR (cm-1): 1612.1, 1550.6 (C=C); 1H-NMR: δ 4.85 (s, 2H, CH2), 7.27-7.30 (m, 3H, Ph-3H), 7.44 (d, J=6.9 Hz, 2H, Ph-2H), 8.27-8.36 (m, 2H, phthalazinyl-2H), 8.63 (d, J=7.9 Hz, 1H, phthalazinyl-H), 8.79 (d, J=7.9 Hz,1H, phthalazinyl-H).
1-Chloro-4-phenylthiomethylphthalazine (7b)
Prepared using 6b as described for 7a, 105.7 g, (84%), m.p. 173-175 oC; MS: m/z 321, 323 (MH+); IR (cm-1): 1619.2, 1583.6, 1512.1 (C=C); 1H-NMR: δ 4.84 (s, 2H, CH2), 7.25 (b, 1H, Ph-H), 7.55-7.64 (m, 2H, Ph-2H), 8.29-8.37 (m, 2H, phthalazinyl-2H), 8.72 (d, J=7.9 Hz, 1H, phthalazinyl-H), 8.68 (d, J=7.9 Hz,1H, phthalazinyl-H).
1-(4-Fluoro-3-trifluoromethylanilino)-4-phenylthiomethylphthalazine (9)
A mixture of 7a (0.86 g, 3 mmol) and 8a (0.72 g, 4 mmol) in isopropanol (20 mL) was heated to 50 oC for 3 h, then the mixture was concentrated in vacuo. The resulting red oil was triturated with diethyl ether (30 mL), by stirring for 10 min to give a suspension. Filtration and recrystallization from ethyl acetate/cyclohexane yielded 1.0 g (81%) of 9, m.p. 214-215 oC; MS: m/z 430 (MH+); IR (cm-1): 3440.3 (NH), 1613.7, 1556.6, 1506.9 (C=C); 1H-NMR: δ 4.84 (s, 2H, CH2), 7.25-7.31 (m, 3H, Ar1-3H), 7.40 (d, J=6.9 Hz, 2H, Ar1-2H), 7.69 (t, J=6.8 Hz, 1H, Ar2-H), 7.97 (m, 1H, Ar2-H), 8.14 (m, 1H, Ar2-H), 8.28 (m, 2H, phthalazinyl-2H), 8.52 (m, 1H, phthalazinyl-H), 8.96 (m, 1H, phthalazinyl-H); Anal. Calcd. for C22H15F4N3S: C 61.53, H 3.52, N 9.79; Found: C 61.42, H 3.41, N 9.70.
1-(3,5-Difluoroanilino)-4-phenylthiomethylphthalazine (10)
Prepared using 7a and 8b as described for 9, 0.97 g, (85%), m.p. 198-199 oC; MS: m/z 380 (MH+); IR (cm-1): 3441.8 (NH), 1625.8, 1553.9, 1478.6 (C=C); 1H-NMR: δ 4.88 (s, 2H, CH2), 7.12 (b, 1H, Ar2-H), 7.26 (t, J 7.4 Hz, 1H, Ar1-H), 7.32 (t, J=7.5 Hz, 2H, Ar1-2H), 7.40 (d, J=7.5 Hz, 2H, Ar1-2H), 7.55 (d, J=7.8 Hz, 2H, Ar2-2H), 8.20-8.26 (m, 2H, phthalazinyl-2H), 8.50 (d, J=8.0 Hz, 1H, phthalazinyl-H), 8.95 (d, J 8.0 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C21H15F2N3S: C 66.48, H 3.98, N 11.07; Found: C 66.52, H 3.79, N 11.12.
1-(4-Trifluoromethoxyanilino)-4-(3,4-difluorophenylthiomethyl)phthalazine (11)
Prepared using 7b and 8c as described for 9, 1.2 g, (84%), m.p. 219-221 oC; MS: m/z 464 (MH+); IR (cm-1): 3465.6 (NH), 1601.7, 1548.0, 1506.6 (C=C); 1H-NMR: δ 4.85 (s, 2H, CH2), 7.23 (m, 1H, Ar2-H), 7.33-7.42 (q, J=8.8 Hz, 1H, Ar2-H), 7.52 (d, J=8.6 Hz, 2H, Ar2-2H), 7.60-7.67 (m, 1H, Ar1-H), 7.75 (d, J=8.6 Hz, 2H, Ar1-2H), 8.22-8.26 (m, 2H, phthalazinyl-2H), 8.47-8.49 (b, 1H, phthalazinyl-H), 9.04-9.05 (b, 1H, phthalazinyl-H); Anal. Calcd. for C22H14F5N3OS: C 57.02, H 3.04, N 9.07; Found: C 57.13, H 3.15, N 9.10.
1-(3-Chloro-4-fluoroanilino)-4-(3,4-difluorophenylthiomethyl)phthalazine (12)
Prepared using 7b and 8d as described for 9, 1.1 g, (87%), m.p. 208-210 oC; MS: m/z 432 (M+); IR (cm-1): 3442.1 (NH), 1616.3, 1565.4, 1505.0 (C=C); 1H-NMR: δ 4.86 (s, 2H, CH2), 7.12 (b, 1H, Ar2-H), 7.36-7.41 (q, J=8.6 Hz, 1H, Ar1-H), 7.59 (t, J=9.0 Hz, 1H, Ar1-H), 7.62-7.65 (m, 2H, Ar2-2H), 7.96 (d, J=5.2 Hz, 1H, Ar1-H), 8.21-8.26 (m, 2H, phthalazinyl-2H), 8.48 (d, J=7.4 Hz, 1H, phthalazinyl-H), 9.02 (d, J=7.3 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C21H13ClF2N3S: C 58.40, H 3.03, N 9.73; Found: C 58.40, H 2.89, N 9.58.
1-(4-Fluoro-3-trifluoromethylanilino)-4-(3,4-difluorophenylthiomethyl)phthalazine (13)
Prepared using 7b and 8a as described for 9, 1.1 g, 80%, m.p. 220-222 oC; MS: m/z 466 (MH+); IR (cm-1): 3447.2 (NH), 1604.2, 1564.8, 1503.5 (C=C); 1H-NMR: δ 4.86 (s, 2H, CH2), 7.21-7.23 (b, 1H, Ar1-H), 7.35-7.42 (q, J=8.5 Hz, Ar2-H), 7.59-7.70 (q, J=8.5 Hz, 2H, Ar1-2H), 7.99 (m, 1H, Ar2-H), 8.17 (d, J=7.8 Hz, 1H, Ar2-H), 8.21-8.25 (m, 2H, phthalazinyl-2H), 8.47-8.50 (m, 1H, phthalazinyl-H), 8.97 (b, 1H, phthalazinyl-H); Anal. Calcd. for C22H13F6N3S: C 56.77, H 2.82, N 9.03; Found: C 56.61, H 2.90, N 8.98.
1-(3,5-Dichloroanilino)-4-(3,4-difluorophenylthiomethyl)phthalazine (14)
Prepared using 7b and 8e as described for 9, 1.0 g, 87%, m.p. 233-234 oC; MS: m/z 448 (M+); IR (cm-1): 3437.8 (NH), 1605.7, 1571.7, 1502.1 (C=C); 1H-NMR: δ 4.88 (s, 2H, CH2), 7.22 (d, J 8.6 Hz, 1H, Ar1-H), 7.36-7.40 (q, J=8.6 Hz, 1H, Ar1-H), 7.47 (s, 1H, Ar2-H), 7.63 (t, J=8.2 Hz, 1H, Ar1-H), 7.89 (s, 2H, Ar2-2H), 8.19-8.24 (m, 2H, phthalazinyl-2H), 8.48 (d, J=7.8 Hz, 1H, phthalazinyl-H), 8.90 (d, J=7.8 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C21H13Cl2F2N3S: C 56.26, H 2.92, N 9.37; Found: C 56.37, H 2.83, N 9.38.
1-(4-Trifluoromethoxyanilino)-4-phenylthiomethyl)phthalazine (15)
Prepared using 7a and 8c as described for 9, 1.1 g, 85%, m.p. 193-195 oC; MS: m/z 428 (MH+); IR (cm-1): 3447.9 (NH), 1619.0, 1555.8, 1508.2 (C=C); 1H-NMR: δ 4.84 (s, 2H, CH2), 7.24-7.33 (m, 3H, Ar1-3H), 7.40 (d, J=7.7 Hz, 2H, Ar1-H), 7.50 (d, J=8.5 Hz, 2H, Ar2-2H), 7.77 (d, J=8.5 Hz, 2H, Ar2-2H), 8.23-8.26 (m, 2H, phthalazinyl-2H), 8.49 (d, J=8.0 Hz, 1H, phthalazinyl-H), 9.05 (d, J=8.0 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C22H16F3N3OS: C 61.28, H 3.77, N 9.83; Found: C 61.99, H 3.56, N 9.63.
1-(4-Fluoro-3-trifluoromethylanilino)-4-(3,4-difluorophenylsulfinylmethyl)phthalazine (16)
30% aqueous H2O2 (2.2 g, 19 mmol) was added into a solution of 13 (6.0 g, 13 mmol) in acetic acid (10 mL).The reaction mixture was stirred for 16 h at room temperature. It was then poured into water and neutralized with 5% NaOH. Filtration and recrystallization from ethyl acetate/chloroform gave 5.7 g (91%) of 16, m.p. 233-234 oC; MS: m/z 482 (MH+); IR (cm-1): 3355.2 (NH), 1574.4, 1505.2, (C=C); 1H-NMR: δ 4.78-4.99 (q, J=13.2 Hz, 2H, CH2), 7.49-7.63 (m, 3H, Ar1-3H), 7.77 (t, J=9.0 Hz, 1H, Ar2-H), 8.03 (m, 2H, Ar2-2H), 8.25 (s, 2H, phthalazinyl-2H), 8.44 (d, J=8.1 Hz, 1H, phthalazinyl-H), 8.57 (d, J=8.1 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C22H13F6N3OS: C 54.89, H 2.72, N 8.73; Found: C 55.03, H 2.93, N 8.95.
1-(3,5-Dichloroanilino)-4-(3,4-difluorophenylsulfinylmethyl)phthalazine (17)
Prepared using 14 as described for 16, 5.8 g, (96%), m.p. 241-242 oC. MS: m/z 465 (MH+); IR (cm‑1): 3425.9 (NH), 1592.4, 1575.8, 1502.5 (C=C); 1H-NMR: δ 4.83-5.01 (q, J=6.7 Hz, 2H, CH2), 7.22 (s, 1H, Ar2-H), 7.46 (d, J=6.1 Hz, 1H, Ar1-H), 7.59-7.63 (q, J=8.4 Hz, 1H, Ar1-H), 7.77 (t, J=7.9 Hz, 1H, Ar1-H), 7.99 (s, 2H, Ar2-2H), 8.10 (s, 2H, phthalazinyl-2H), 8.33 (d, J=8.0 Hz, 1H, phthalazinyl-H), 8.58 (d, J 8.0 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C21H13Cl2F2N3OS: C 54.32, H 2.82, N 9.05; Found: C 54.29, H 2.73, N 9.16.
1-(4-Trifluoromethoxyanilino)-4-phenylsulfinylmethylphthalazine (18)
Prepared using 15 as described for 16, 5.0 g, 91%, m.p. 225-226 oC. MS: m/z 466 (M+Na+); IR (cm-1): 3438.5 (NH), 1637.2, 1560.6, 1509.6 (C=C); 1H-NMR: δ 4.79-4.83 (q, J=6.8 Hz, 2H, CH2), 7.34 (d, J=8.9 Hz, 2H, Ar2-2H), 7.50-7.53 (m, 3H, Ar1-3H), 7.64 (d, J=7.7 Hz, 2H, Ar1-2H), 7.99 (d, J =8.0 Hz, 2H, Ar2-2H), 8.05 (s, 2H, phthalazinyl-2H), 8.21 (d, J=7.9 Hz, 1H, phthalazinyl-H), 8.63 (d, J =7.9 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C22H16F3N3O2S: C 59.59, H 3.64, N 9.48; Found: C 59.66, H 3.90, N 9.67.
1-(4-Trifluoromethoxyanilino)-4-phenylsulfonylmethylphthalazine (19)
NaWO4·2H2O (2.5 mmol) and 30% aqueous H2O2 (11.3 g, 100 mmol) were added into a solution of 15 (4.5 g, 10.6 mmol) in methanol (20 mL). The mixture was stirred at room temperature for 18 h, filtered and washed with water. Recrystallization from ethyl acetate/acetone afforded 19 as off-white powder, 4.1 g (84%), m.p. 228-229 oC; MS: m/z 460 (MH+); IR (cm-1): 3416.7 (NH), 1622.1, 1566.6, 1508.8 (C=C); 1H-NMR: δ 5.33 (s, 2H, CH2), 7.37 (d, J=8.5 Hz, 2H, Ar2-2H), 7.58 (t, J=7.4 Hz, 2H, Ar1-2H), 7.69-7.76 (m, 3H, Ar1-3H), 7.96 (d, J=8.5 Hz, 2H, Ar2-2H), 7.99 (s, 2H, phthalazinyl-2H), 8.27 (d, J=8.0 Hz, 1H, phthalazinyl-H), 8.60 (d, J=8.1 Hz, 1H, phthalazinyl-H); Anal. Calcd. for C22H16F3N3O3S: C 57.51, H 3.51, N 9.15; Found: C 57.20, H 3.34, N 8.99.
Pharmacology
The anticancer activities of compounds 9-19 were evaluated in vitro on Bel-7402 (Human Liver Cancer cell lines) and HT-1080 (Human Fibro Sarcoma cell lines) by measuring cell viability by the MTT method, with cisplatin as the positive control. The cells were seeded in RPM I 1640 medium (100 μL) in a 96-well plate at a concentration of 4000 cells per well. After culturing for 12 h at 37 oC and 5% CO2, cells were incubated with various concentrations of the samples for 24 h. MTT was added at a terminal concentration of 5μg/mL and incubated with the cells for 4 h. The formazan crystals were dissolved in DMSO (100 μL) in each well and the optical density was measured at 492 nm (for the absorbance of MTT formazan) and 630 nm (for the reference wavelength). The IC50 was calculated using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software.
References
- Tsoungas, P. G.; Searcey, M. A convenient access to benzo-substituted phthalazines as potential precursors to DNA intercalators. Tetrahedron Lett. 2001, 42, 6589–6592. [Google Scholar]
- Sivakumar, R.; Gnanasam, S. K.; Ramachandran, S.; Leonard, J. T. Pharmacological evaluation of some new 1-substituted-4-hydroxyphthalazines. Eur. J. Med. Chem. 2002, 37, 793–801. [Google Scholar]
- Napoletano, M.; Norcini, G.; Pellacini, F.; Marchini, F.; Morazzoni, G.; Ferlenga, P.; Pradella, L. Phthalazine PDE4 Inhibitors. Part 2: the synthesis and biological evaluation of 6-methoxy-1,4-disubstituted derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 33–37. [Google Scholar]
- Haack, T.; Fattori, R.; Napoletano, M.; Pellacini, F.; Fronza, G.; Raffaini, G.; Ganazzoli, F. Phthalazine PDE IV inhibitors: conformational study of some 6-methoxy-1,4-disubstituted derivatives. Bioorg. Med. Chem. 2005, 13, 4425–4433. [Google Scholar] [CrossRef]
- Joule, J. A.; Smith, G. F. Synthesis of diazine rings. Hetercycl. Chem. 1978, 138–139. [Google Scholar]
- Yale, H. L. o-Acetobenzoic acid, its preparation and lactonization. A novel application of the Doebner synthesis. J. Amer. Chem. Soc. 1947, 69, 1547–1548. [Google Scholar]
- Jaques, J.; Marquet, A. Selective α-bromination of an aralkyl ketone with phenyltrimethyl-ammonium tribromide: 2-bromoacetyl-6-methoxynaphthalene and 2,2-dibromoacetyl-6-methoxy-naphthalene. Org. Syn. 1976, 53, 111. [Google Scholar]
- André, B. C.; Carl, B.; David, S. M. An expedient approach to E,Z-dienes using the Julia olefination. Tetrahedron Lett. 2001, 42, 5149–5153. [Google Scholar]
- Sample availability: Samples of the compounds mentioned are available from the corresponding author.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Li, J.; Zhao, Y.-F.; Yuan, X.-Y.; Xu, J.-X.; Gong, P. Synthesis and Anticancer Activities of Novel 1,4-Disubstituted Phthalazines. Molecules 2006, 11, 574-582. https://doi.org/10.3390/11070574
AMA Style
Li J, Zhao Y-F, Yuan X-Y, Xu J-X, Gong P. Synthesis and Anticancer Activities of Novel 1,4-Disubstituted Phthalazines. Molecules. 2006; 11(7):574-582. https://doi.org/10.3390/11070574
Chicago/Turabian StyleLi, Juan, Yan-Fang Zhao, Xiao-Ye Yuan, Jing-Xiong Xu, and Ping Gong. 2006. "Synthesis and Anticancer Activities of Novel 1,4-Disubstituted Phthalazines" Molecules 11, no. 7: 574-582. https://doi.org/10.3390/11070574