Salicylanilide Acetates: Synthesis and Antibacterial Evaluation

Abstract
:Introduction
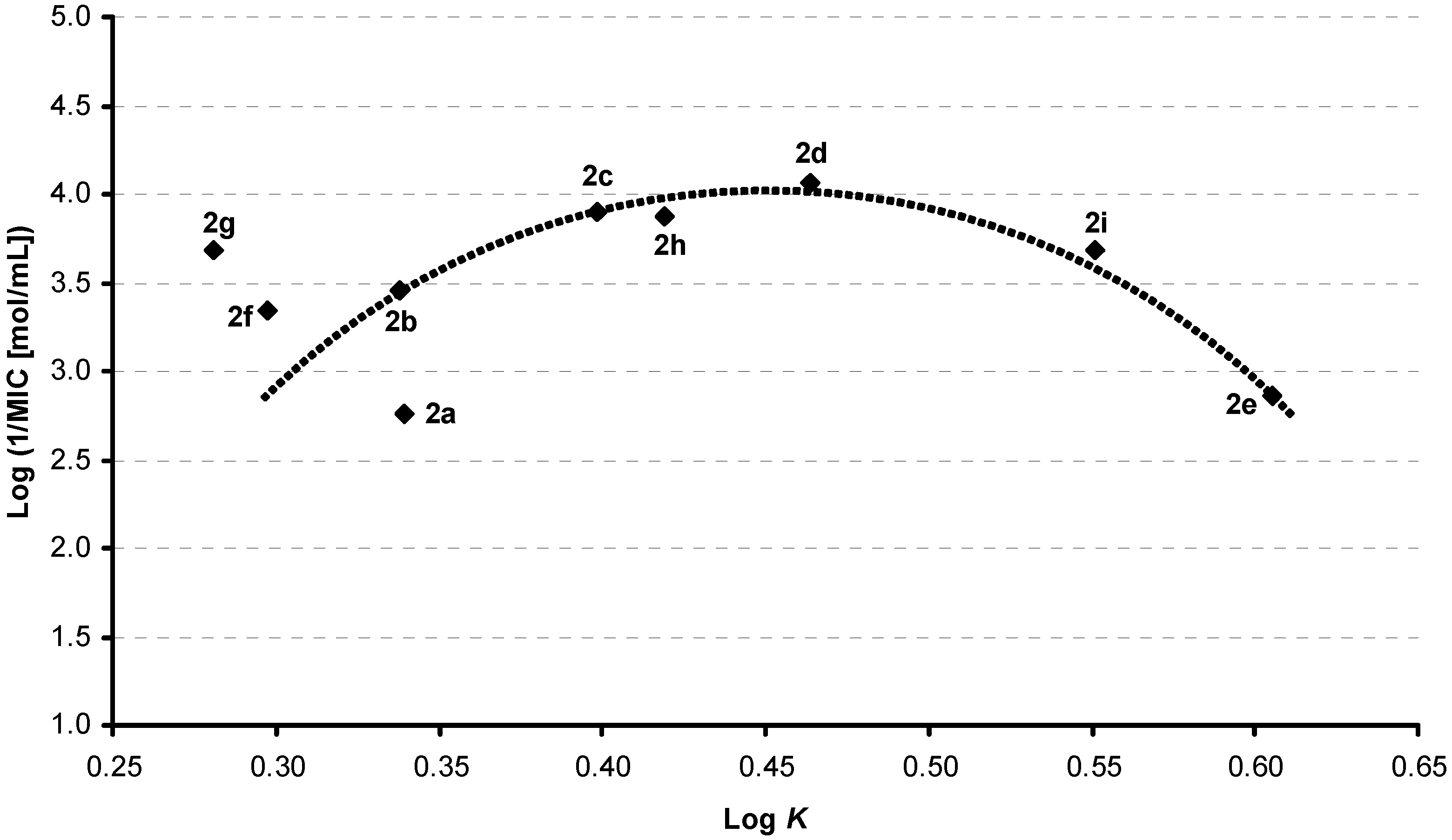
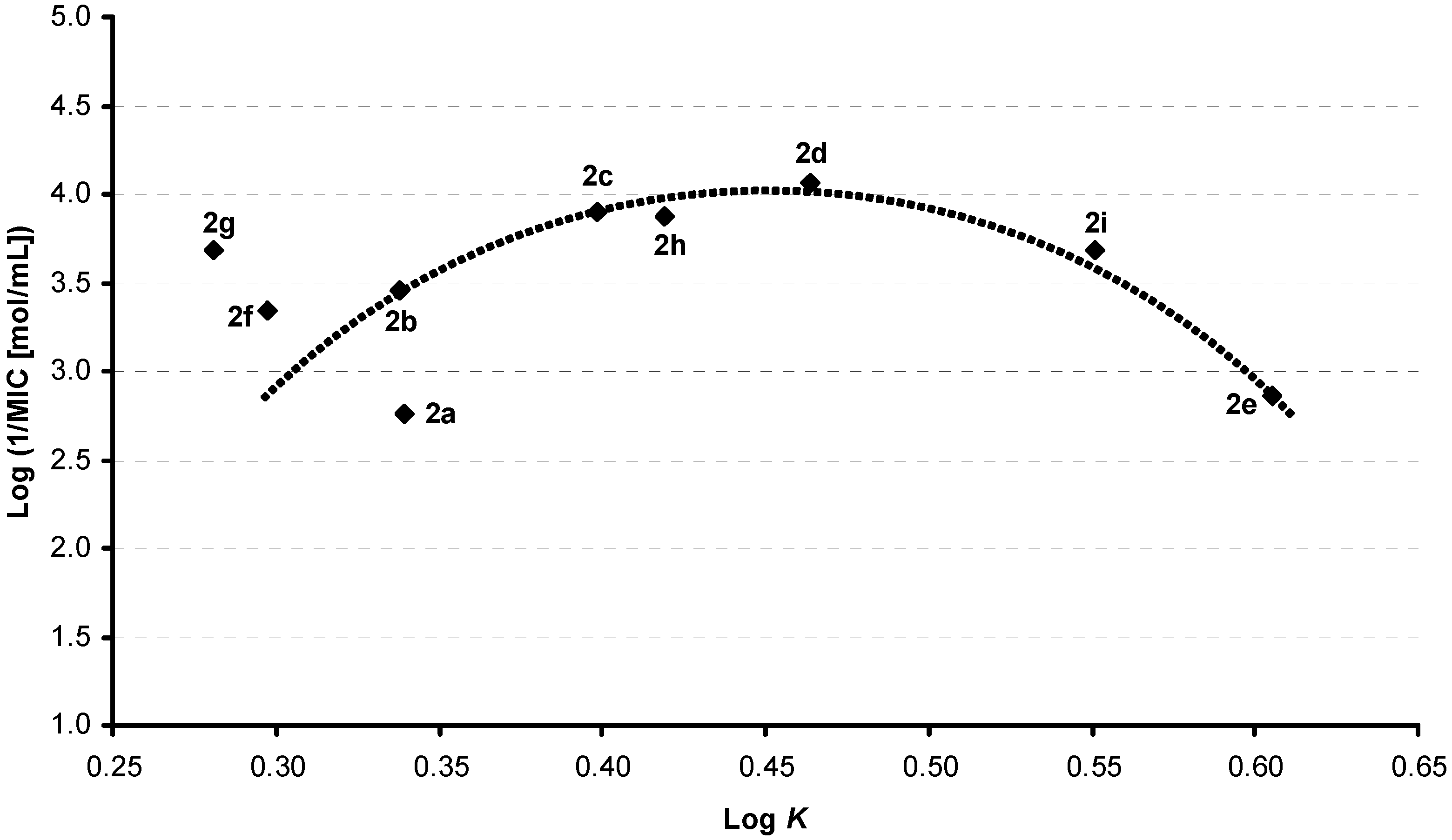
Results and Discussion
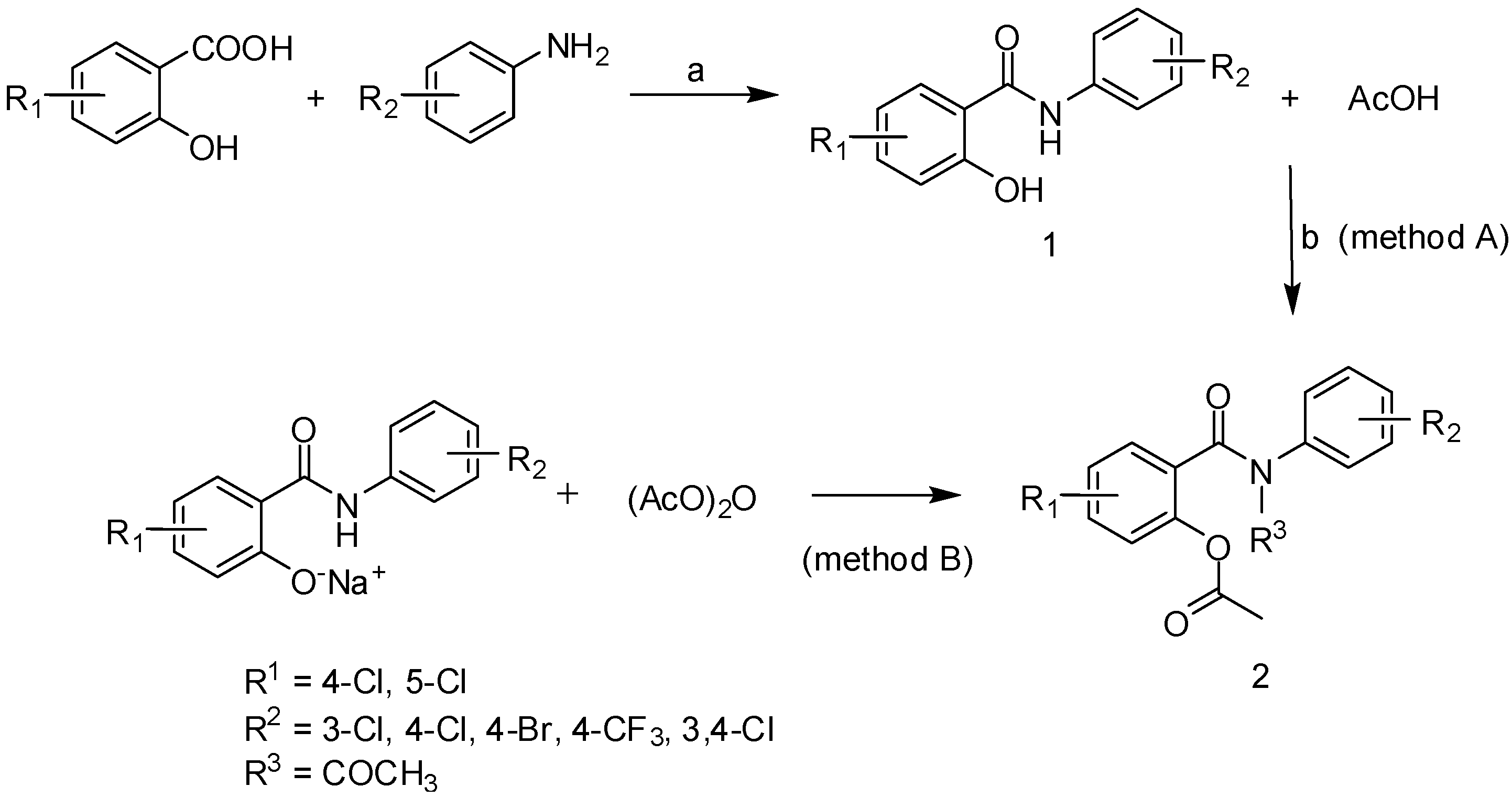

{kind=link}
{kind=link}
 | |||||
| Comp. | R1 | R2 | log K | log P/ClogP ChemOffice | log P ACD/Log P |
| 1a | 5-Cl | 3-Cl | 0.4698 | 3.57 / 5.08085 | 5.44 ± 0.42 |
| 1b | 5-Cl | 4-Cl | 0.4021 | 3.57 / 5.08085 | 5.40 ± 0.42 |
| 1c | 5-Cl | 4-Br | 0.4927 | 3.84 / 5.23085 | 5.58 ± 0.49 |
| 1d | 5-Cl | 4-CF3 | 0.5580 | 3.93 / 5.44405 | 5.43 ± 0.46 |
| 1e | 5-Cl | 3,4-Cl | 0.6558 | 4.12 / 5.75025 | 6.31 ± 0.44 |
| 1f | 4-Cl | 3-Cl | 0.3004 | 3.57 / 5.08085 | 5.39 ± 0.42 |
| 1g | 4-Cl | 4-Cl | 0.2953 | 3.57 / 5.08085 | 5.35 ± 0.42 |
| 1h | 4-Cl | 4-CF3 | 0.4225 | 3.93 / 5.44405 | 5.37 ± 0.46 |
| 1i | 4-Cl | 3,4-Cl | 0.6132 | 4.12 / 5.75025 | 6.25 ± 0.44 |
 | ||||||
| Comp. | R1 | R2 | R3 | log K | log P/ClogP ChemOffice | log P ACD/Log P |
| 2a | 4-Cl | 3-Cl | H | 0.3391 | 3.54 / 3.87285 | 3.46 ± 0.40 |
| 2b | 4-Cl | 4-Cl | H | 0.3377 | 3.54 / 3.87285 | 3.42 ± 0.40 |
| 2c | 4-Cl | 4-Br | H | 0.3983 | 3.81 / 4.02285 | 3.59 ± 0.44 |
| 2d | 4-Cl | 4-CF3 | H | 0.4637 | 3.90 /4.23605 | 3.44 ± 0.42 |
| 2e | 4-Cl | 3,4-Cl | H | 0.6053 | 4.10 / 4.5004 | 4.32 ± 0.41 |
| 2f | 5-Cl | 3-Cl | H | 0.2971 | 3.54 / 3.87285 | 3.74 ± 0.40 |
| 2g | 5-Cl | 4-Cl | H | 0.2806 | 3.54 / 3.87285 | 3.70 ± 0.40 |
| 2h | 5-Cl | 4-CF3 | H | 0.4188 | 3.90 / 4.23605 | 3.72 ± 0.42 |
| 2i | 5-Cl | 3,4-Cl | H | 0.5510 | 4.10 / 4.5004 | 4.60 ± 0.41 |
| 2j | 4-Cl | 4-Br | COCH3 | 0.3117 | 3.76 / 3.83856 | 3.17 ± 0.63 |
| Comp. | MIC [µg/mL] | |||||||
| CA | CT | CK | CG | TB | AF | AC | TM | |
| 24 h | 24 h | 24 h | 24 h | 24 h | 24 h | 24 h | 72 h | |
| 48 h | 48 h | 48 h | 48 h | 48 h | 48 h | 48 h | 120 h | |
| 2a | 31.25 | 31.25 | 7.81 | 62.5 | 15.63 | 7.81 | 7.81 | 0.98 |
| 250 | 250 | 62.5 | 500 | 500 | 7.81 | 7.81 | 0.98 | |
| 2b | 31.25 | 31.25 | 15.63 | 15.63 | 15.63 | 7.81 | 15.63 | 1.95 |
| 125 | 15.63 | 15.63 | 15.63 | 15.63 | 7.81 | 15.63 | 1.95 | |
| 2c | 7.81 | 7.81 | 3.91 | 15.63 | 7.81 | 7.81 | 7.81 | 0.98 |
| 15.63 | 7.81 | 3.91 | 15.63 | 7.81 | 7.81 | 7.18 | 0.98 | |
| 2d | 7.81 | 3.91 | 3.91 | 3.91 | 3.91 | 7.81 | 3.91 | 0.49 |
| 7.81 | 7.81 | 3.91 | 7.81 | 7.81 | 7.81 | 7.81 | 0.49 | |
| 2e | 31.25 | 31.25 | 31.25 | 31.25 | 62.5 | 7.81 | 250 | 0.98 |
| 62.50 | 62.50 | 31.25 | 250 | 250 | 31.25 | 250 | 0.98 | |
| 2f | 31.25 | 7.81 | 31.25 | 15.63 | 15.63 | 15.63 | 15.63 | 1.95 |
| 62.50 | 31.25 | 62.50 | 62.50 | 62.50 | 15.63 | 15.63 | 1.95 | |
| 2g | 15.63 | 15.63 | 7.81 | 15.63 | 3.91 | 15.63 | 7.81 | 0.89 |
| 31.25 | 31.25 | 7.91 | 15.63 | 7.81 | 15.63 | 15.63 | 0.98 | |
| 2h | 7.81 | 15.63 | 3.91 | 15.63 | 7.81 | 3.91 | 7.81 | 0.49 |
| 15.63 | 15.63 | 3.91 | 15.63 | 7.81 | 3.91 | 7.81 | 0.49 | |
| 2i | 3.91 | 15.63 | 1.95 | 3.91 | 7.81 | 15.63 | 7.81 | 0.98 |
| 7.81 | 31.25 | 3.91 | 3.91 | 31.25 | 62.50 | 7.81 | 0.98 | |
| 2j | ≤ 31.25 | 31.25 | 31.25 | 31.25 | ≤ 31.25 | ≤ 31.25 | ≤ 31.25 | ≤ 31.25 |
| 31.25 | 31.25 | 31.25 | 62.50 | ≤ 31.25 | ≤ 31.25 | ≤ 31.25 | ≤ 31.25 | |
| FLU | 0.06 | 0.12 | 3.91 | 0.98 | 0.24 | >125 | >125 | 1.95 |
| 0.12 | >125 | 15.62 | 3.91 | 0.48 | >125 | >125 | 3.91 | |
| Comp. | M. tuberculosis My 331/88 | M. avium My 330/88 | M. kansasii My 235/80 | M. kansasii My 6500/96 | Cytotoxicity IC50 [μg/mL] | ||||
| 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | ||
| 2a | 8 | 8 | 16 | 16 | 8 | 8 | 8 | 16 | 9.82 |
| 2b | 2 | 2 | 8 | 8 | 4 | 8 | 4 | 4 | 5.19 |
| 2c | 2 | 2 | 8 | 8 | 4 | 8 | 4 | 8 | 60.60 |
| 2d | 4 | 4 | 4 | 4 | 2 | 2 | 2 | 4 | 12.60 |
| 2e | 1 | 1 | 8 | 8 | 4 | 4 | 4 | 4 | 0.82 |
| 2f | 4 | 4 | 8 | 16 | 8 | 8 | 8 | 8 | 7.25 |
| 2g | 4 | 4 | 8 | 8 | 8 | 16 | 8 | 16 | 7.74 |
| 2h | 4 | 4 | 8 | 8 | 2 | 4 | 4 | 4 | 0.27 |
| 2i | 2 | 2 | 8 | 8 | 4 | 4 | 4 | 4 | 1.18 |
| 2j | 4 | 4 | 8 | 8 | 4 | 8 | 4 | 8 | 1.23 |
| INH | 0.5 | 1 | >250 | >250 | >250 | >250 | 4 | 4 | >100 |
Conclusions
Experimental
General
HPLC determination of lipophilicity (capacity factor K / calculated log K)
Lipophilicity calculations
Synthesis of salicylanilide acetates 2a-j
Antifungal evaluation
Antimycobacterial evaluation
Cytotoxicity assay
Acknowledgements
References
- Vinsova, J.; Imramovsky, A. Salicylanilides: Still a Topical Potential Antibacterially active Group. Ces. Slov. Farm. 2004, 53, 294–299. [Google Scholar]
- Hassan, G. S.; Hegazy, G. H.; Safwat, H. M. Synthesis of Furo-salicylanilides and Their Heterocyclic Derivatives with Anticipated Molluscicidal Activity. Arch. Pharm. Chem. Life Sci. 2006, 339, 448–455. [Google Scholar] [CrossRef]
- Deng, W.; Guo, Z.; Guo, Y.; Feng, Z.; Jiang, Y.; Chu, F. Acryolylamino-salicylanilides as EGFR PTK inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 469–472. [Google Scholar]
- Liechti, Ch.; Séquin, U.; Bold, G.; Furet, P.; Meyer, T.; Traxler, P. Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor. Eur. J. Med. Chem. 2004, 39, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Hlasta, D. J.; Demers, J. P.; Foleno, B. D.; Fraga-Spano, S. A.; Guan, J.; Hilliard, J. J.; Macielag, M. J.; Ohemeng, K. A.; Sheppard, C. M.; Sui, Z.; Webb, G. C.; Weidner-Wells, M. A.; Barret, J. F. Novel inhibitors of bacterial two-component systems with gram positive antibacterial activity: Pharmacophore identification based on the screening hit closantel. Bioorg. Med. Chem. Lett. 1998, 8, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Macielag, M. J.; Demers, J. P.; Fraga-Spano, S. A.; Hlasta, D. J.; Johnson, S. G.; Kanojia, R. M.; Russell, R. K.; Sui, Z.; Weidner-Wells, M. A.; Werblood, H.; Foleno, B. D.; Goldschmidt, R. M.; Loeloff, M. J.; Webb, G.C.; Barrett, J. F. Substituted Salicylanilides as Inhibitors of Two-Component Regulatory Systems in Bacteria. J. Med. Chem. 1998, 41, 2939–2945. [Google Scholar]
- Imramovsky, A.; Vinsova, J.; Ferriz, J. M.; Kunes, J.; Pour, M.; Dolezal, M. Salicylanilide esterification: unexpected formation of novel seven-membered rings. Tetrahedron Lett. 2006, 47, 5007–5511. [Google Scholar]
- Waisser, K.; Hladuvkova, J.; Kunes, J.; Kubicova, L.; Klimesova, V.; Karajannis, P.; Kaustova, J. Synthesis and antimycobacterial activity of salicylanilides substituted in position 5. Chem. Pap. 2001, 55, 121–129. [Google Scholar]
- Dolezal, M.; Palek, L.; Vinsova, J.; Buchta, V.; Jampilek, J.; Kralova, K. Substituted Pyrazinecarboxamides: Synthesis and Biological Evaluation. Molecules 2006, 11, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Jampilek, J.; Buchta, V.; Silva, L.; Niedbala, H.; Podeszwa, B.; Palka, A.; Majerz-Maniecka, K.; Oleksyn, B.; Polanski, J. Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar] [CrossRef] [PubMed]
- Vinsova, J.; Cermakova, K.; Tomeckova, A.; Ceckova, M.; Buchta, V.; Jampilek, J.; Cermak, P.; Kunes, J.; Dolezal, M.; Staud, F. Synthesis and antimicrobial evaluation of new 2-substituted 5,7-di-tert-butylbenzoxazoles. Bioorg. Med. Chem. 2006, 14, 5850–5865. [Google Scholar] [CrossRef] [PubMed]
- Ciampa, G.; Tricko, C. Rend. Accad. Sci. Fis. Naples 1966, 33, 385, [Chem. Abstr. 1968, 68, 95469].
- National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeast: Approved Standard, NCCLS document, M27-A; NCCLS: Villanova, PA, U.S.A, 1997. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival – Application to proliferation and cyto-toxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Vinsova, J.; Imramovsky, A.; Buchta, V.; Ceckova, M.; Dolezal, M.; Staud, F.; Jampilek, J.; Kaustova, J. Salicylanilide Acetates: Synthesis and Antibacterial Evaluation. Molecules 2007, 12, 1-12. https://doi.org/10.3390/12010001
Vinsova J, Imramovsky A, Buchta V, Ceckova M, Dolezal M, Staud F, Jampilek J, Kaustova J. Salicylanilide Acetates: Synthesis and Antibacterial Evaluation. Molecules. 2007; 12(1):1-12. https://doi.org/10.3390/12010001
Chicago/Turabian StyleVinsova, Jarmila, Ales Imramovsky, Vladimir Buchta, Martina Ceckova, Martin Dolezal, Frantisek Staud, Josef Jampilek, and Jarmila Kaustova. 2007. "Salicylanilide Acetates: Synthesis and Antibacterial Evaluation" Molecules 12, no. 1: 1-12. https://doi.org/10.3390/12010001