Synthesis of Functionalised Nucleosides for Incorporation into Nucleic Acid-Based Serine Protease Mimics

Ghent University, Department of Organic Chemistry, Laboratory for Organic and Biomimetic Chemistry, Krijgslaan 281 (S4), B-9000 Gent, Belgium
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(1), 114-129; https://doi.org/10.3390/12010114
Submission received: 21 December 2006
/
Revised: 29 January 2007
/
Accepted: 30 January 2007
/
Published: 31 January 2007
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The synthesis of nucleosides modified with an extra imidazole, carboxyl and hydroxyl group is described. These nucleosides can be incorporated into an oligonucleotide duplex, thus generating a novel type of serine protease mimic.
Introduction
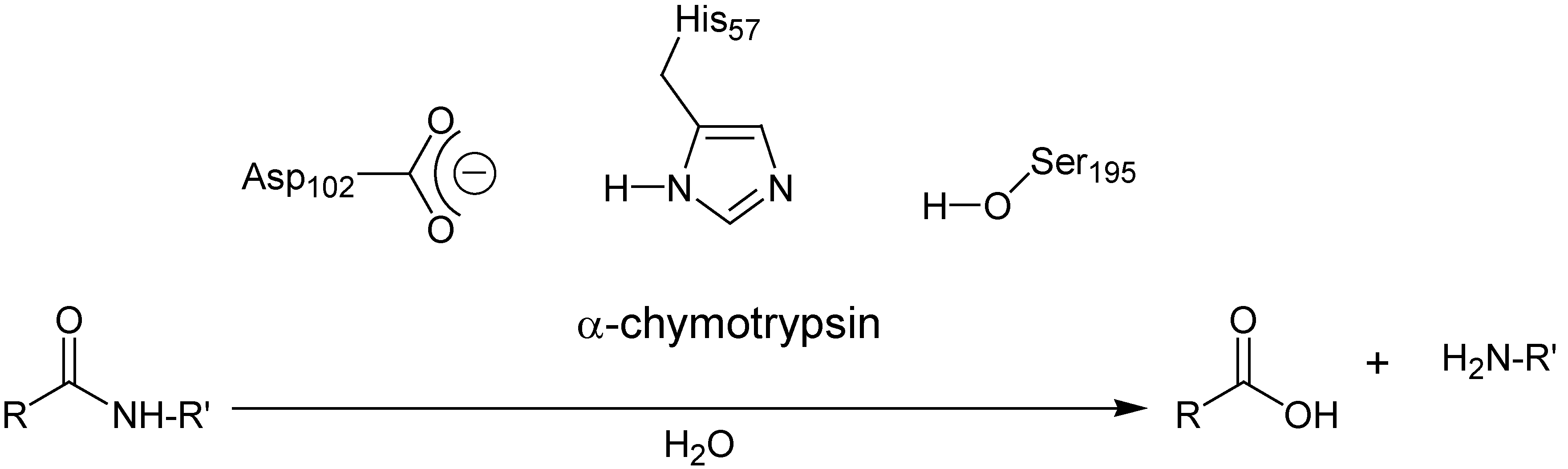
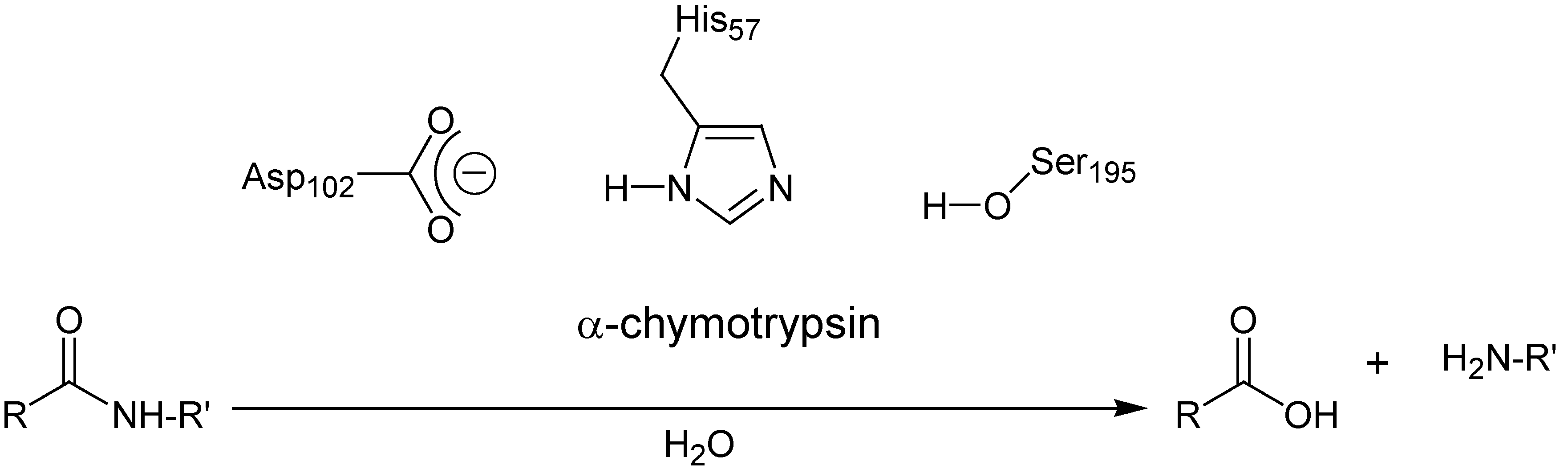
Enzymes are of great importance in nature because of their ability to catalyse reactions in a selective and efficient way. The development of artificial catalysts which mimic enzymatic activity has always been a very challenging area of research. Among the different enzymes, proteases have been intensively studied because of their unequalled capacity for hydrolysing amide and peptide bonds under mild conditions. One of the most studied serine proteases is α-chymotrypsin, and X-ray diffraction studies give a good insight into the tertiary structure of the enzyme [1,2,3,4]. The active site contains three amino acids which are essential for catalysis of the cleavage of amide bonds (Figure 1). The hydrolysis is realized by the unique collaboration between Histidine-57 (His-57), Serine-195 (Ser-195) and Aspartate-102 (Asp-102) [5].
Figure 1.
The catalytic triad of α-chymotrypsin for peptide hydrolysis.
Intrigued by this performance, several groups have tried to construct model systems with hydrolytic activity and different peptide based systems have been developed in this context [6,7,8,9,10,11,12,13,14,15,16]. We must bear in mind that stable secondary structures such as helices only become important for rather long sequences of polypeptides. Therefore, synthetic polymers, synzymes and modified cyclodextrins have also been studied as possible catalytic systems [17,18,19,20,21]. Although a big variety of artificial hydrolases has been synthesized, none have even approached the efficiency of α-chymotrypsin.
Two approaches have been used in our laboratory to develop potentially catalytic hydrolytic systems. In a first approach, we developed non-peptidic organic molecules containing an array of functional groups in a suitable geometry for possible hydrolytic activity [22]. In a second combinatorial approach, a dipodal scaffold based on a cholic acid template was synthesized and two independent peptide chains were attached, each containing one residue of the catalytic triad [23,24]. In addition, a model based on a tripodal scaffold, with a rigid structure possessing three independent functionalised peptide chains, was also synthesized [25].
We recently became interested in the development of a totally new class of model systems based on nucleic acid building blocks. A major drawback in using oligonucleotides in the design of synthetic catalysts is that they lack functional groups with such diversity and properties as proteins in order to act as bases or Lewis acids. However, it is tempting to equip oligonucleotides with the functional tools of peptide chains by building block modification. This should result in a powerful combination of both predefined structural organization (duplex formation) and presence of catalytic entities.
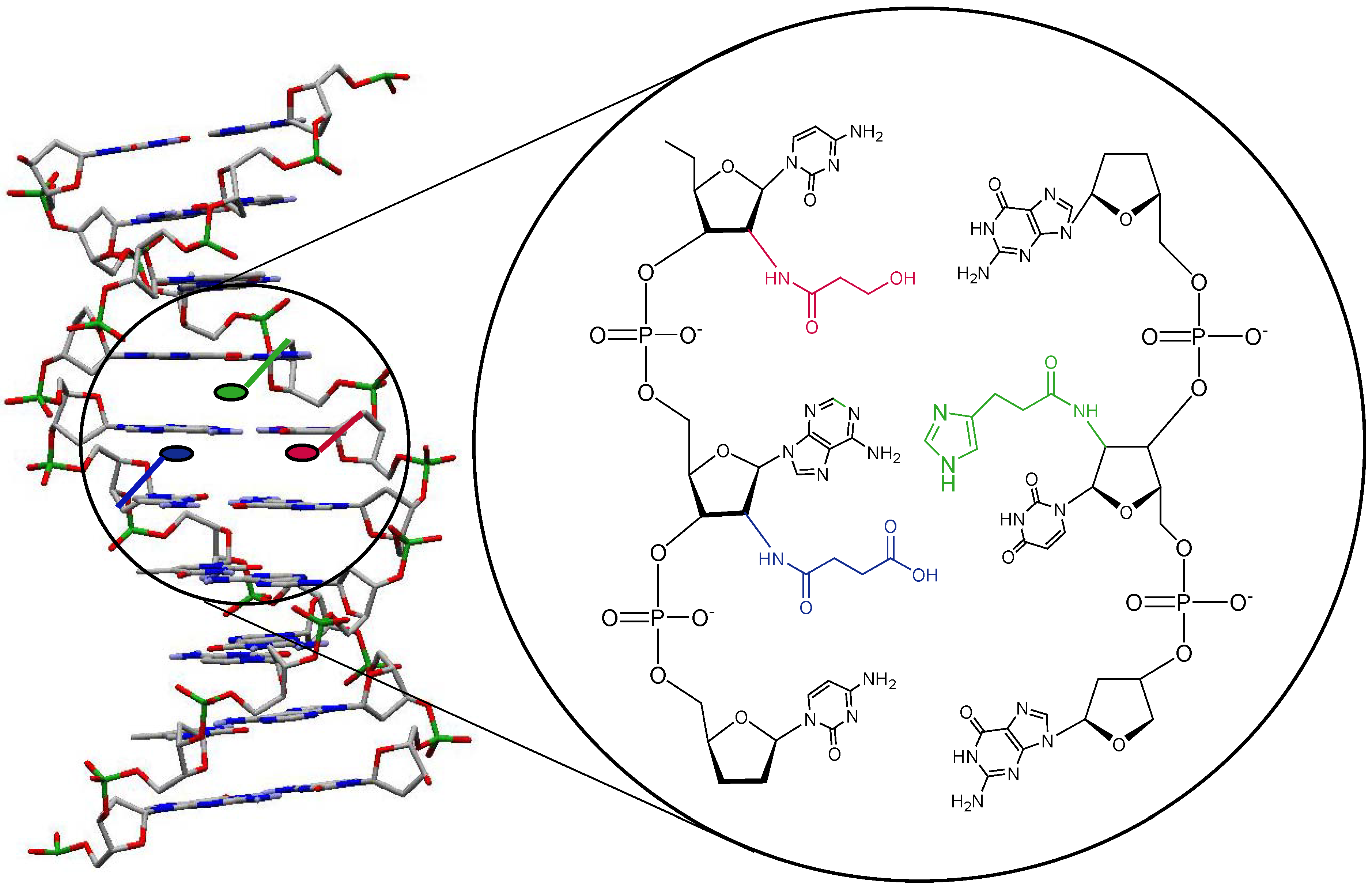
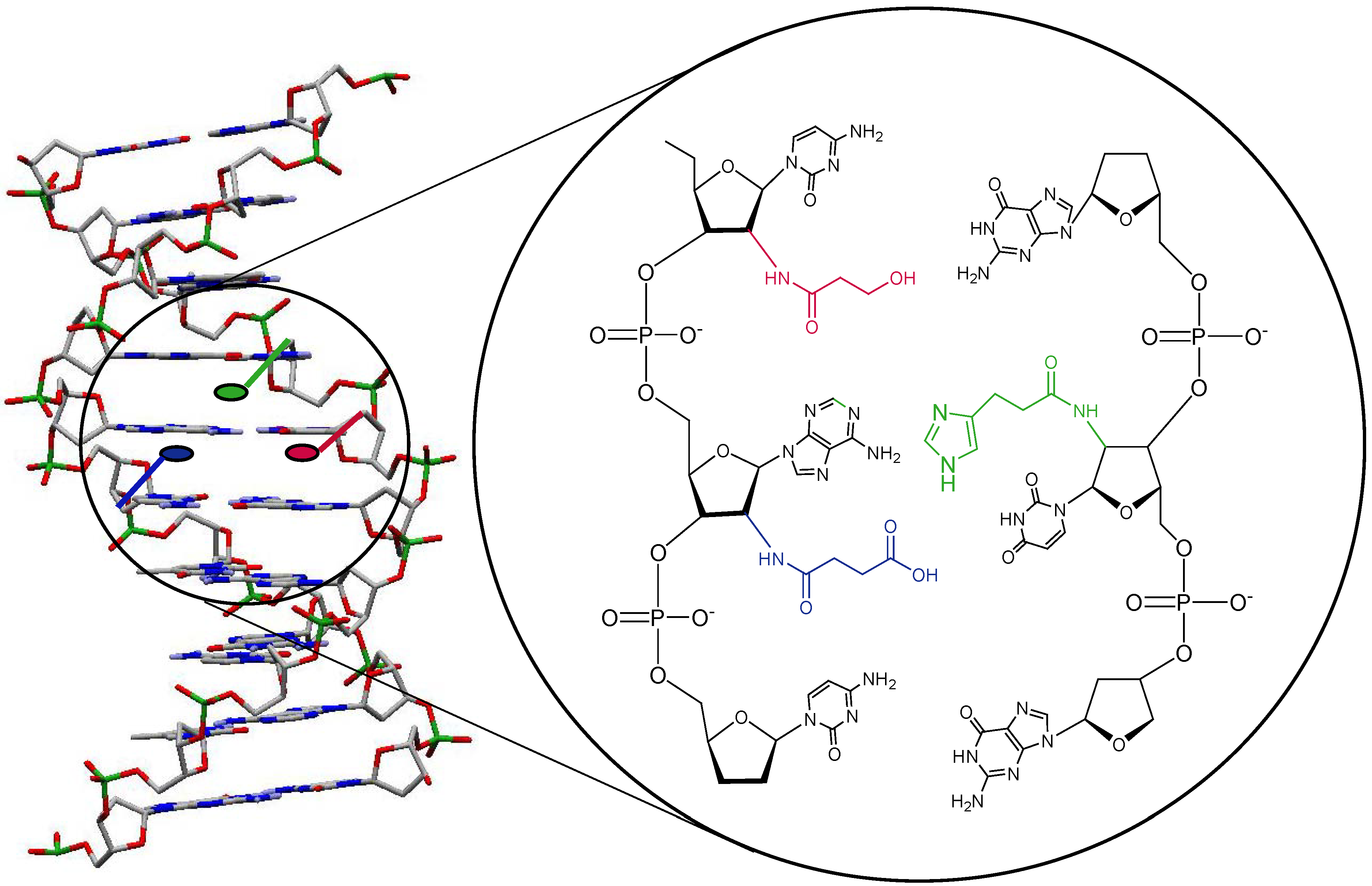
More specifically, we envisaged the creation of an active site via duplex formation between two complementary sugar modified oligonucleotide strands (Figure 2). The cleavage of the target peptide bonds will occur via functionalities in the minor and major groove of the DNA duplex. Both strands of the duplex will have several different functional group modifications. As for the catalytic groups, imidazole, alcohol and carboxylate groups would be included in accordance with the functional groups in the active site of serine proteases. Depending on the specific site of incorporation of the functionalities in the nucleoside building blocks, different spatial positions within the duplex grooves can be reached. In a first stage we have chosen to synthesize a series of functionalised nucleosides starting from 2’-amino-2’-deoxyuridine.
Figure 2.
Creation of an active site.
Since DNA synthesis has become a reliable and relatively simple automated process, the synthesis of modified ribo- and deoxyribooligonucleotides is a standard procedure [26]. After the synthesis of suitably modified nucleosides, a wide variety of oligonucleotides can be synthesized in an automated way.
Results and Discussion
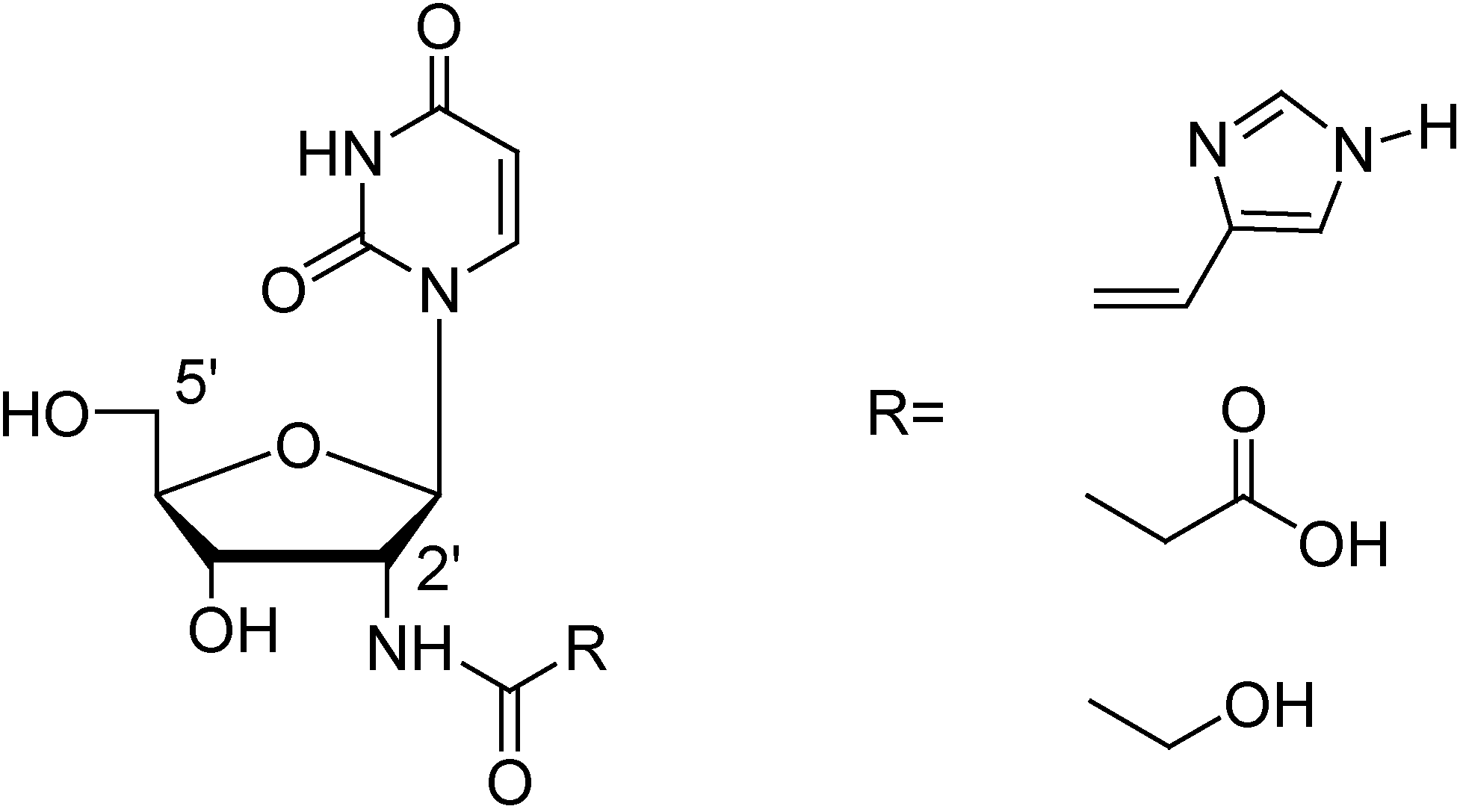
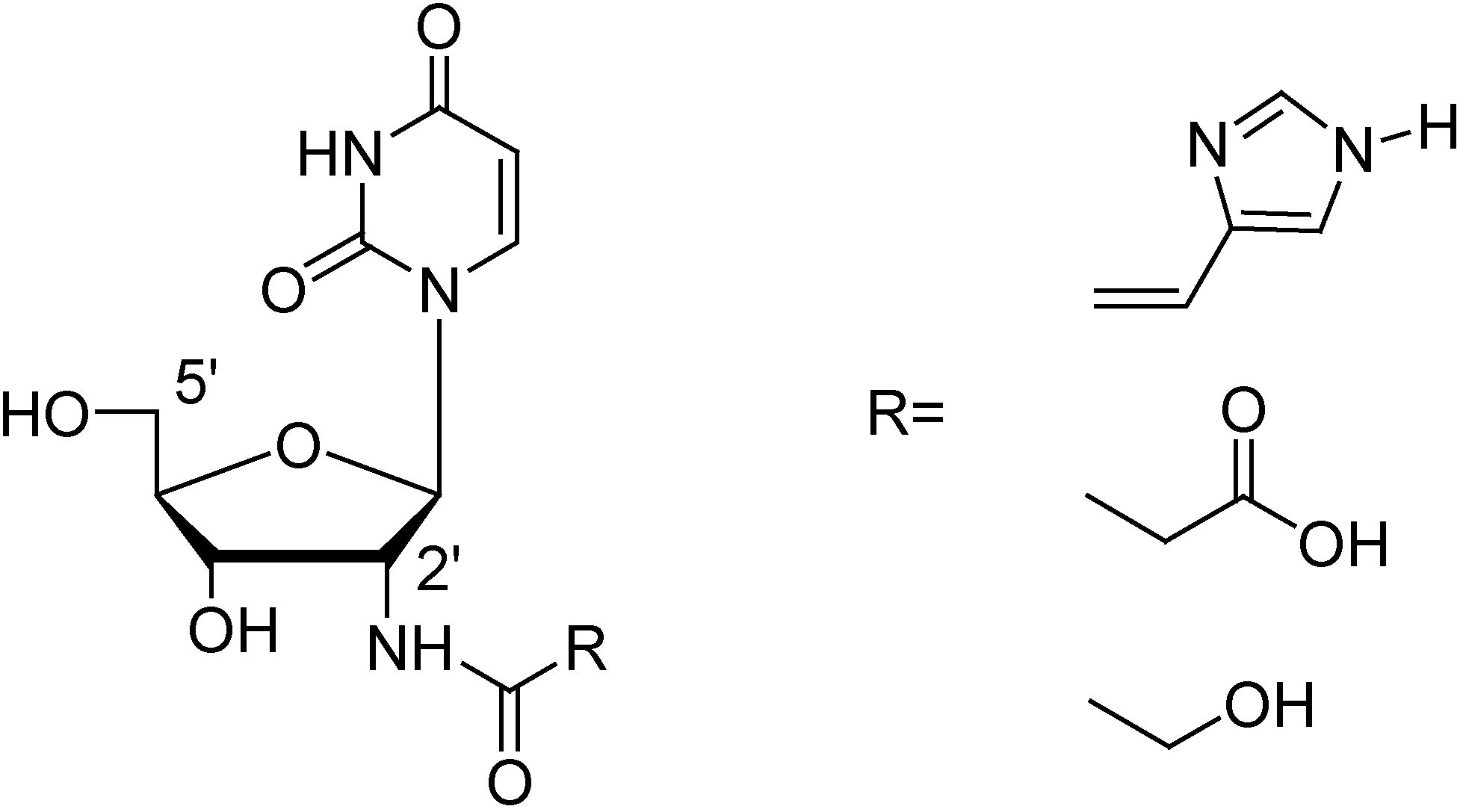
Figure 3 shows the envisaged building blocks. Starting from 2’-amino-2’-deoxyuridine, prepared according to literature procedures, three different functional groups would be attached via an amide bond [27,28].
Figure 3.
2’-Amido modified building blocks.
An important aspect when synthesizing functionalised oligonucleotides is the appropriate protection of the extra functionalities on the corresponding nucleoside building blocks. Not only should these protecting groups survive the DNA synthesis conditions, but they should also be stable under all chemical conditions used for construction of the required suitably protected phosphoramidites.
Modification of oligonucleotides with an imidazole functionality has been described before. To circumvent known problems with imidazole protection during nucleoside and oligonucleotide synthesis, precursor-based techniques employing post-synthetic functionalization have been developed. The imidazole moiety is introduced after oligonucleotide synthesis either at the 5’-end or internally, using a base-modified building block [29,30]. Post-synthetic conjugation to a 2’-amino building block has been reported, but only using a long linker [31].
We chose to introduce the imidazole functionality in the nucleoside and to incorporate it into oligonucleotides. As for imidazole containing phosphoramidite building blocks, the imidazole moiety has been mostly connected to the nucleoside via the heterocyclic base. No phosphoramidite building blocks containing an imidazole moiety on C 2’, connected via an amide bond, have been previously described.
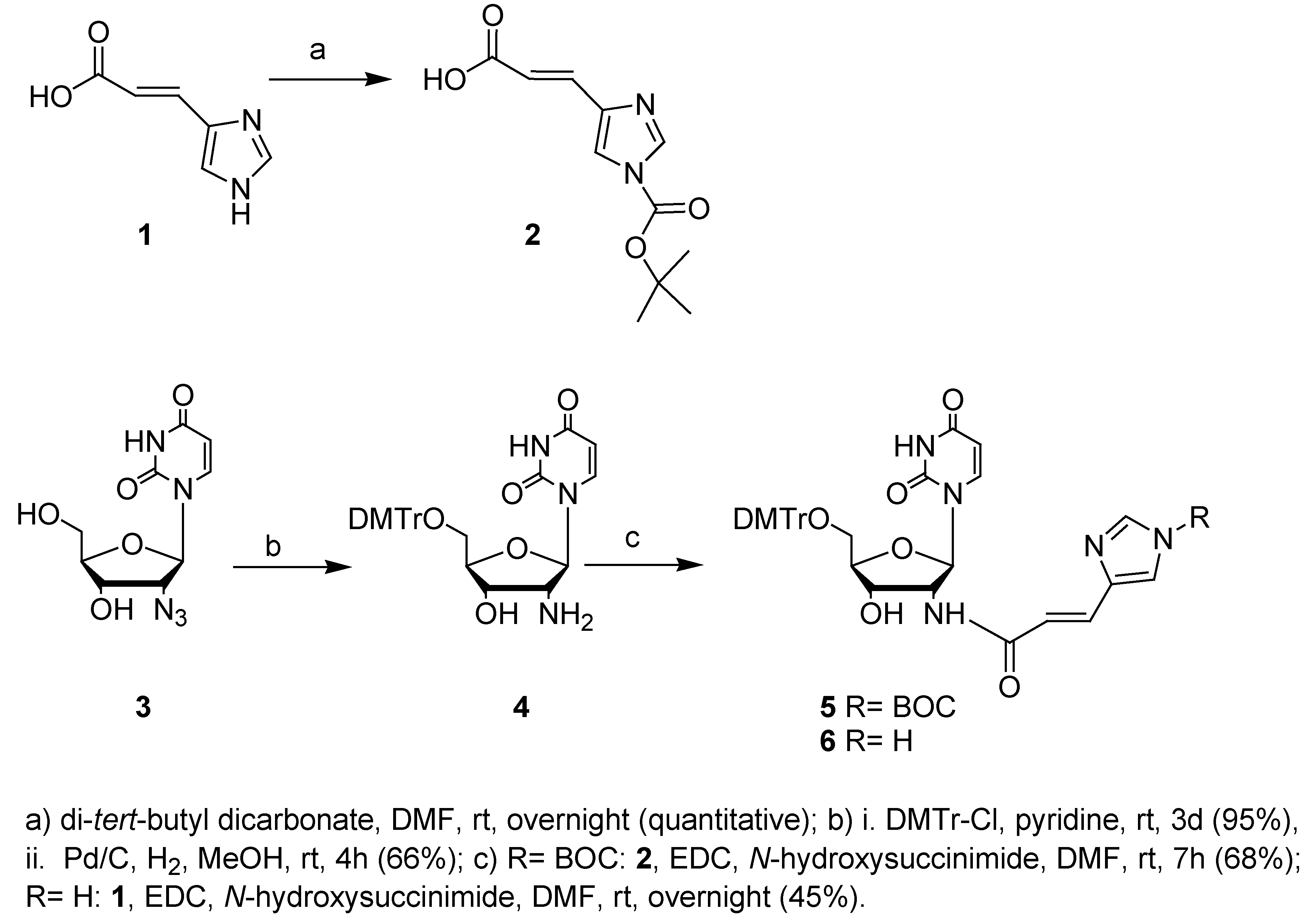
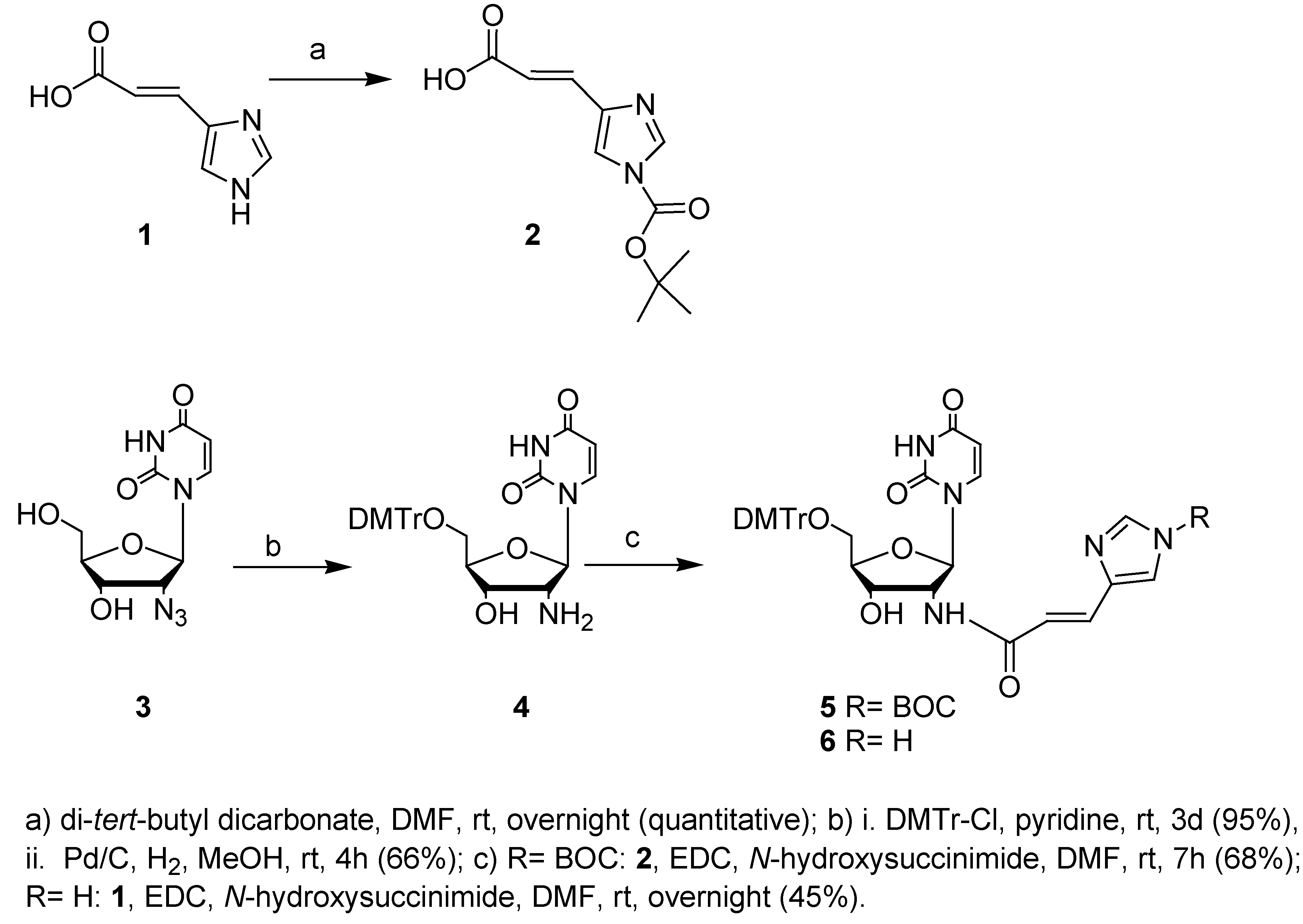
The use of a 2,4-dinitrophenyl group (DNP), which has been reported as a protecting group for the imidazole functionality of His in peptide chemistry and in stepwise solid phase synthesis of nucleopeptides, was our first choice [32,33]. However, the DNP protection did not survive conditions used during purification of the final DMTr-protected nucleoside. Because it has further been proven that the DNP group gives yellow contaminated products after deprotection, we decided to change the protecting group into a tert-butoxycarbonyl (Boc) one [34], which is compatible with the reagents used during nucleoside- and oligonucleotide synthesis [35,36,37]. In principle, removal of the Boc group on a solid support can be accomplished with a 10% TFA solution after automated synthesis [37]. However, it has been shown that such a TFA treatment can lead to a certain amount of depurination of the oligonucleotide [34,38]. The research groups of Gait and Herdewijn both proved that removal of the Boc group from imidazole during standard deprotection procedures with saturated methanolic ammonia after oligonucleotide synthesis is possible [35,39], therefore in our second attempt, using urocanic acid (1), the imidazole moiety was Boc-protected using di-tert-butyldicarbonate as shown in Scheme 1.
NIm-Boc-protected urocanic acid 2 was further coupled with the dimethoxytrityl protected 2’-amino-2’-deoxyuridine 4 in the presence of N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide (EDC) and N-hydroxysuccinimide (NHS) to give 5 in 68% yield. Coupling of the Boc-protected urocanic acid 2 with unprotected 2’-amino-2’-deoxyuridine followed by dimethoxytritylation of this amide also gave the desired compound 5, albeit in lower yield. The dimethoxytrityl protected amine 4 was synthesized starting from the known 2’-azido-2’-deoxyuridine (3) [27,28]. Selective dimethoxytritylation of the 5’-hydroxyl function, followed by reduction of the azide gave us the dimethoxytrityl protected amine 4 in good yield.
Since incorporation of unprotected imidazole building blocks into oligonucleotides has been accomplished in some cases, we also directly coupled urocanic acid (1) to dimethoxytrityl protected 2’-amino-2’-deoxyuridine 4 [40,41]. This was achieved using EDC and NHS, yielding the desired compound 6 in only moderate yield [42,43]. Alternative coupling procedures using N,N’-dicyclo-hexylcarbodiimide (DCC) and NHS, N,N’-diisopropylcarbodiimide (DIC) and polystyrene-bound 1-hydroxybenzotriazole (HOBt) or the pentafluorophenol ester of urocanic acid (1) were not effective.
Scheme 1.
Synthesis of imidazole-modified nucleoside building blocks 5 and 6.
Base and sugar modification of nucleosides with a carboxylic acid has been described and protection of the carboxylate function proved to be necessary for oligonucleotide synthesis. As for our envisaged carboxylic acid building block, shown in Scheme 2, a derivative similar to 11, only differing in the carboxylic acid protecting group, has been synthesized before. However, this modified nucleoside was incorporated only at the end of oligonucleotide sequences [44]. The carboxyl group was protected as a benzyl ester, which can be selectively cleaved by hydrogenolysis. However, the use of hydrogen gas leads to significant reduction of double bonds in the pyrimidine bases and the final modified oligonucleotide was adsorbed on charcoal of the Pd catalyst, requiring a catalytic phase transfer procedure using Pd nanoparticles.
Methyl- and ethyl esters have also been used as carboxylic acid protecting groups. Basic hydrolysis of these esters is frequently plagued by low yields, long reaction times, the need for large excesses of reagents, and the formation of by-products that are difficult to separate [45,46,47].
In our quest for a more straightforward alternative avoiding additional deprotection steps, our first choice of carboxylate protecting group was the 9-fluorenylmethyl (Fm) group. This group is often used as a protecting group for Asp in the stepwise solid-phase synthesis of nucleopeptides [48]. This protecting group is stable under the conditions used in oligonucleotide synthesis and can be removed with the standard oligonucleotide deprotection reagents. However, loss of the Fm group was observed under conditions used for purification of the required DMTr-protected nucleoside.
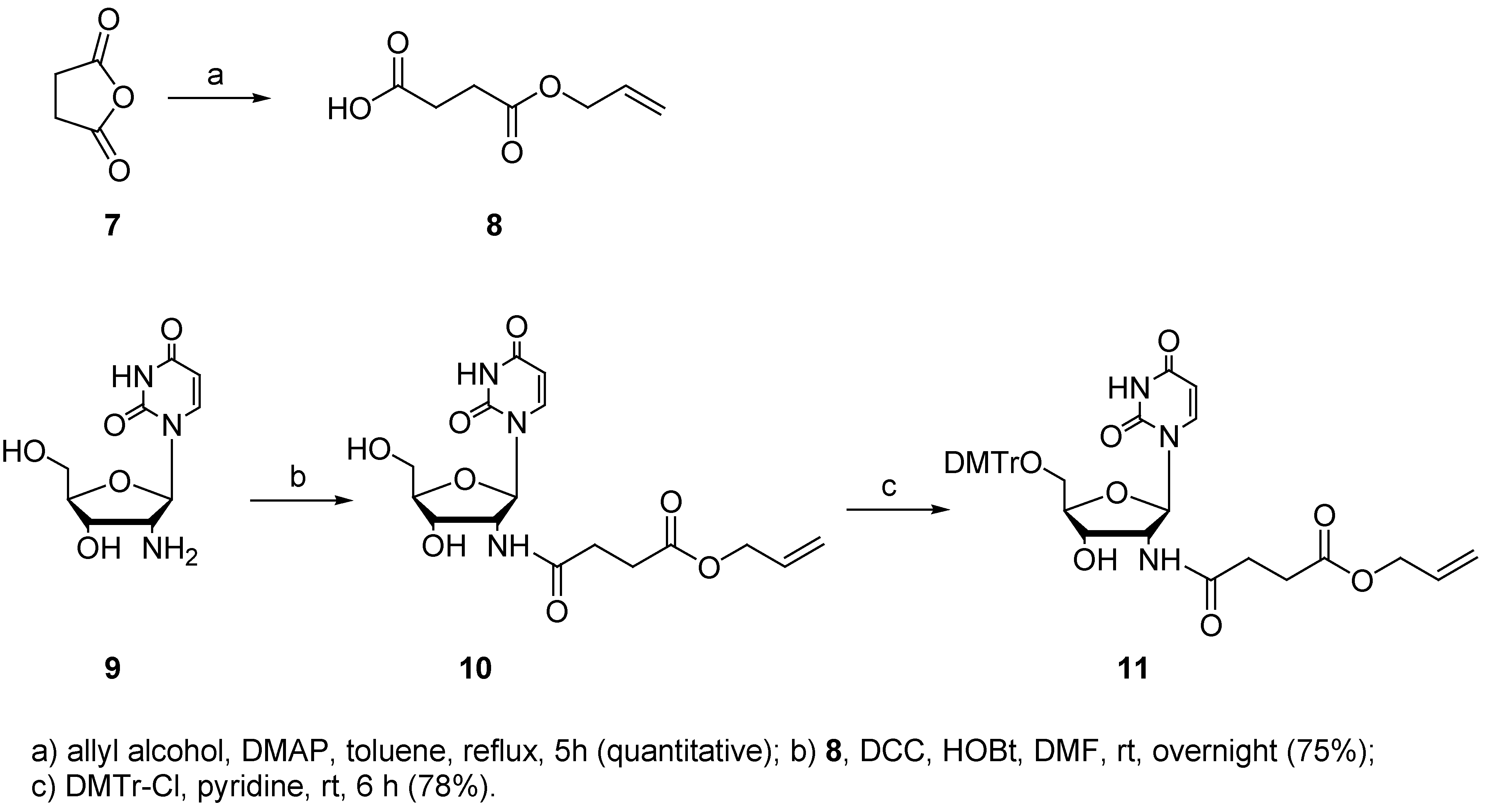
Since for other carboxylate-modified building blocks allyl protection was reported to be removed smoothly, our next choice was the use of an allyl protecting group. This protecting group can be removed after oligonucleotide synthesis by treatment with tetrakis(triphenylphosphine)palladium (0), triphenylphosphine and morpholine [51]. Reaction of allyl alcohol with succinic anhydride (7) in the presence of DMAP afforded the monoprotected dicarboxylic acid 8 [52]. 2’-Amino-2’-deoxyuridine (9) was synthesized from 3 according to literature procedures [27,28]. Coupling of acid 8 with 2’-amino-2’-deoxyuridine (9) in the presence of DCC and HOBt gave the modified nucleoside 10 [50]. Dimethoxytritylation could be accomplished under standard conditions and the corresponding derivative 11 was isolated and purified without problems.
Scheme 2.
Synthesis of the carboxylate-modified nucleoside building block 11.

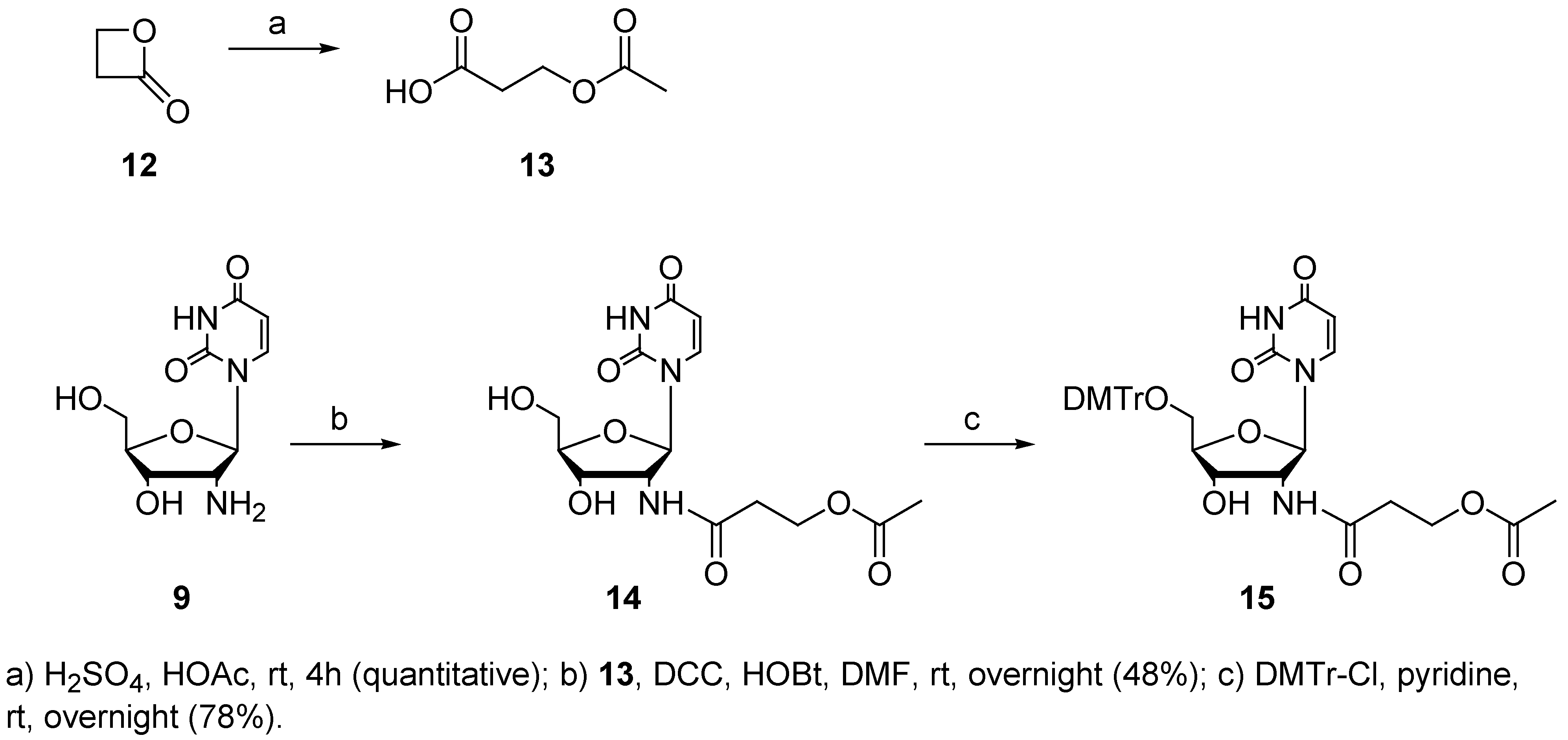
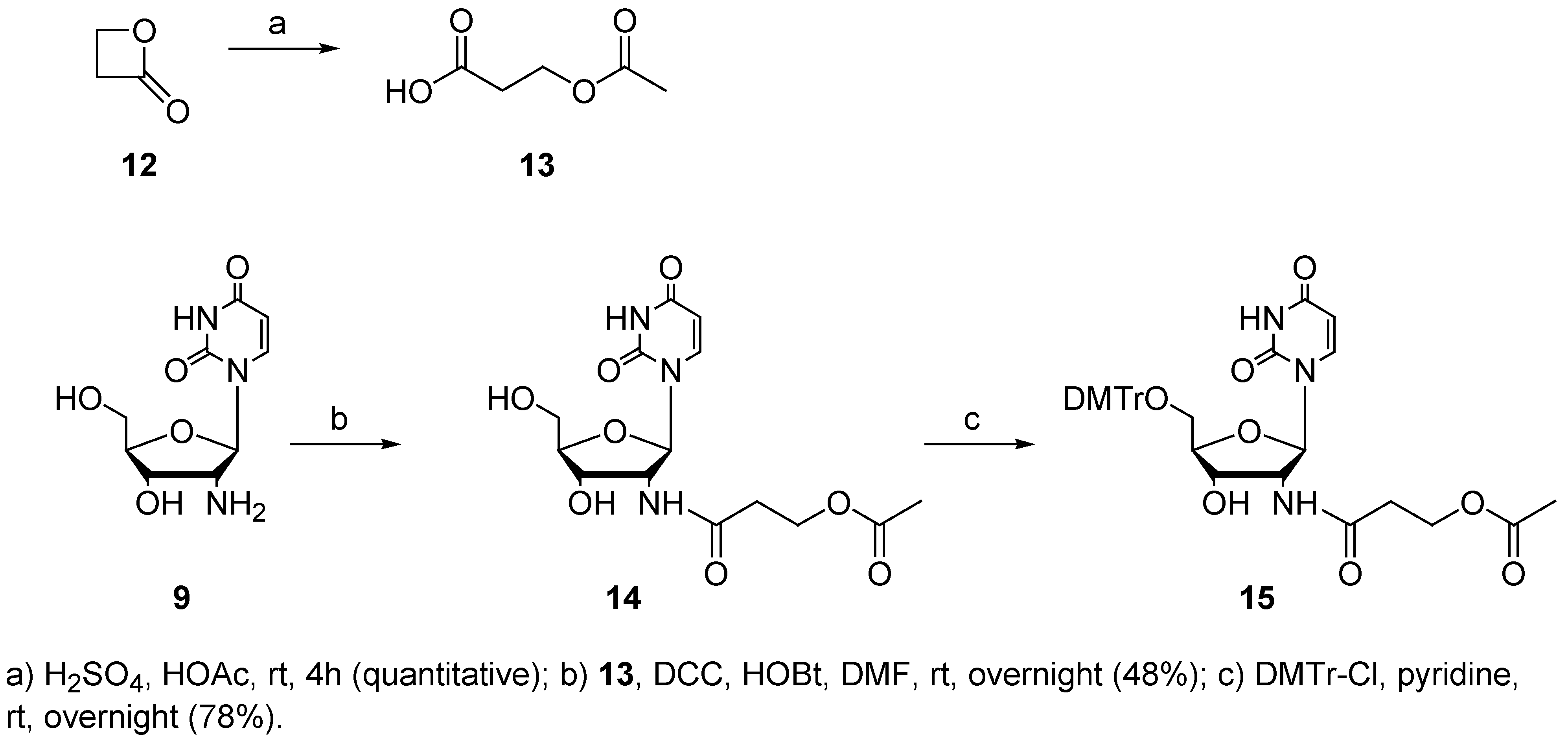
Although oligonucleotides modified with a hydroxyl function on the base, sugar or phosphate backbone have already been synthesized, no previous method for the introduction of an alcohol functionality via a C 2’-amide bond has been reported [53,54,55,56,57,58,59]. Out of the wide variety of available protecting groups for the protection of this hydroxyl function [53,54,55,56,57], we chose the acetate group. Deprotection is then possible by the standard ammonia treatment which is used to remove the cyanoethyl and base protecting groups with concurrent cleavage of the oligonucleotide from the solid phase. Reaction of acetic acid with β-propiolactone (12) afforded protected carboxylic acid 13 [60]. As shown in Scheme 3, carboxylic acid 13 was subsequently coupled with 2’-amino-2’-deoxyuridine (9) in the presence of DCC and HOBt [50]. Dimethoxytritylation of the 5’-OH group was accomplished under standard conditions and resulted in amide 15 in satisfactory yield.
Scheme 3.
Synthesis of the hydroxyl-modified nucleoside building block 15.
Conclusions
In summary, three different nucleoside analogues containing protected imidazole, carboxylate and hydroxyl functionalities on the 2’ position of the sugar moiety − compounds 5, 11 and 15 − respectively, have been synthesized. The incorporation of the different nucleoside analogues into oligonucleotides as a new approach towards nucleic acid based serine protease models is currently under investigation.
Experimental Section
General
All reagents were obtained either from Aldrich, ACROS Organics or Fluka and were used without prior purification. Dimethylformamide (DMF) and pyridine were purchased from ACROS (dry over molecular sieves). Toluene was distilled from sodium and MeOH was dried on Mg/I2. Reactions were performed under argon atmospheres. Analytical TLC was carried out on glass plates precoated with silica gel (Merck, 60F-254, 0.25 mm) and compounds were visualized using phosphomolybdic acid (PMA) and KMnO4. Flash chromatography was performed on silica gel (KieselgelMerck, 230-400 mesh, Type 9385, 60 Angström). IR spectra were recorded on a Perkin-Elmer 1600 series FT-IR spectrometer and bands are quoted in cm-1. 1H-NMR and APT spectra were recorded on a Bruker 500 or 300 MHz spectrometer. The deuterated solvents CDCl3, MeOD, DMSO-d6 were obtained from Aldrich. Chemical shifts (δ units) are expressed in parts per million (ppm) relative to TMS and the internal solvent peak was used for calibration. When peak multiplicities are reported, the following abbreviations are used: s, singlet; d, doublet; t, triplet; m, multiplet. Coupling constants (J values) are expressed in Hertz (Hz). The Attached Proton Test (APT) technique was used to assign 13C peaks (C, CH, CH2, CH3). Mass spectra (ES-MS) were recorded on a quadrupole ion trap LC mass spectrometer (Thermo Finnigan MAT LCQ mass spectrometer) equipped with electrospray ionization and EI-MS spectra were acquired on a Hewlett-Packard 5998 A (MS, EI) spectrometer.
2’-Amino-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (4)
2’-Azido-2’-deoxyuridine (3) was coevaporated three times with pyridine and dried overnight under vacuum. To a solution of 3 (200 mg, 0.74 mmol) in pyridine (6 mL) was added 4,4’-dimethoxy-tritylchloride (378 mg, 1.11 mmol) and the reaction mixture was stirred at rt for 3 days. MeOH (100 μL) was added at 0 °C and the reaction mixture was stirred for 10 min. The reaction mixture was evaporated under reduced pressure and EtOAc (10 mL) was added. The organic layer was washed successively with water (10 mL), saturated NaHCO3 (10 mL) and again with water (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH/Et3N, 98:1:1) to afford 2’-azido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (400 mg, 95%): Rf (DCM + 5% MeOH): 0.34; 1H-NMR (500 MHz, CDCl3) δ 7.97 (1 H, d, J= 8.2 Hz), 7.43-7.33 (9 H, m), 6.91 (4 H, d, J= 8.9 Hz), 6.04 (1 H, d, J= 3.0 Hz), 5.42 (1 H, d, J= 8.2 Hz), 4.54 (1 H, dd, J= 6.12 Hz), 4.23 (1 H, dd, J= 3.0, 5.5 Hz), 4.08 (1 H, m), 3.86 (6 H, s), 3.66 (1 H, AB dd, J= 2.41, 11.3 Hz), 3.55 (1 H, AB dd, J= 2.41, 11.3 Hz), 2.80 (1 H, br s), 1.73 (1 H, br s) ppm; APT (CDCl3, 125 MHz) δ 55.2 (CH3), 61.2 (CH2), 67.1 (CH), 69.6 (CH), 83.1 (CH), 87.2 (C), 87.6 (CH), 102.3 (CH), 113.2 (CH), 127.2 (CH), 127.9 (CH), 128.0 (CH), 129.9 (CH), 130.0 (CH), 134.8 (C), 135.0 (C), 139.3 (CH), 144.0 (C), 149.7 (C), 158.6 (C), 162.2 (C) ppm; IR (KBr plate) ν 3500, 2114, 1689 cm-1; ESI-MS (positive mode) 303.3 (DMTr) +, 573.2 (M + Na) +. 2’-Azido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (400 mg, 0.7 mmol) was dissolved in MeOH (7 mL) and Pd/C (32 mg) was added. The flask was equipped with a H2 balloon and the reaction mixture was stirred at room temperature for 4h. The reaction mixture was filtered through Celite® to remove the Pd/C and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH/Et3N, 95:4:1) to afford 4 (250 mg, 66%): Rf (DCM/ MeOH, 95:5) 0.2; 1H-NMR (500 MHz, MeOD) δ 7.81 (1 H, d, J= 8.1 Hz), 7.40 (2 H, d, J= 7.3 Hz), 7.31- 7.28 (6 H, m), 7.23 (1 H, m), 6.87 (4 H, d, J= 8.4 Hz), 5.85 (1 H, d, J= 6.8 Hz), 5.32 (1 H, d, J= 8.1 Hz), 4.28 (1 H, dd, J= 2.93, 5.43 Hz), 4.11 (1 H, m), 3.77 (6 H, s), 3.51 (1 H, dd, J= 5.6, 6.7 Hz); 3.42 (1 H, AB dd, J= 3.3, 10.7 Hz), 3.38 (1 H, AB dd, J= 3.1, 10.1 Hz) ppm; APT (MeOD, 125 MHz) δ 55.8 (CH3), 59.9 (CH), 64.7 (CH2), 72.8 (CH), 79.0 (C), 79.3 (C), 86.4 (CH), 90.4 (CH), 102.9 (CH), 114.2 (CH), 128.1 (CH), 128.9 (CH), 129.4 (CH), 131.4 (CH), 136.6 (C), 142.2 (CH), 145.9 (C), 160.3 (C) ppm; IR (KBr plate) ν 3375, 2970; 2834, 2352; 1865, 1842, 1694, 1651, 1607, 1510, 1462, 1381, 1251, 1176, 1086, 1034, 829, 791, 702 cm-1; ESI-MS (positive mode) 303.1 (DMTr) +, 545.3 (M) +, 567.7 (M + Na) +.
3-(1-tertbutoxycarbonyl-imidazol-4-yl)-acrylic acid (2)
A solution of urocanic acid (1, 1 g, 7.2 mmol) in DMF (25 mL) was stirred for 1h and then di-tert-butyldicarbonate (1.58 g, 7.2 mmol) was added. The reaction mixture was stirred overnight at rt and was then coevaporated with toluene to afford 2 in quantitative yield: 1H-NMR (300 MHz, MeOD) δ 8.27 (1 H, s), 7.94 (1 H, s), 7.46 (1 H, d, J = 15.6 Hz), 6.42 (1 H, d, J = 15.6 Hz), 1.57 (9 H, s) ppm; APT (MeOD, 75 MHz) δ 27.4 (CH3), 85.9 (C), 118.5 (CH), 119.5 (CH), 119.6 (CH), 135.2 (CH), 138.4 (C), 146.3 (C), 167.5 (C) ppm; IR (KBr plate) ν 3144, 2979, 2934, 2802, 2668, 2802, 2668, 2544, 1902, 1758, 1703, 1657, 1563, 1492, 1457, 1384, 1336, 1320, 1302, 1255, 1204, 1148, 989, 841, 656 cm-1; ESI-MS (positive mode) 139.0 (M- Boc)+, 182.8 (M- tBu)+, 238.2 (M) +.
2’-[3-(1-tertbutoxycarbonyl-imidazol-4-yl)acryloyl]-amido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxy-uridine (5)
A solution of 2 (154 mg, 0.65 mmol) in DMF (2 mL) was cooled to 0 °C. 1-(3-Dimethylamino-propyl)-3-ethylcarbodiimide hydrochloride (125 mg, 0.65 mmol) and N-hydroxysuccinimide (75 mg, 0.65 mmol) were added. The mixture was allowed to warm to rt and was stirred overnight. The reaction mixture was then cooled to 0 °C and 4 (300 mg, 0.54 mmol) was added. The reaction was allowed to warm to room temperature and stirred for 7h, then H2O was added and the aqueous layer was extracted with EtOAc (3 x). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH, 97:3) to afford 5 (240 mg, 68%): Rf (DCM/MeOH, 9/1): 0.5; 1H-NMR (300 MHz, MeOD) δ 8.25 (1 H, s), 7.93 (1 H, d, J = 8.3 Hz), 7.74 (1 H, s), 7.52-7.23 (10 H, m), 6.91 (4 H, d, J = 8.9 Hz), 6.79 (1 H, d, J = 15.4 Hz), 6.17 (1 H, d, J= 8.5 Hz), 5.34 (1 H, d, J = 8.1 Hz), 5.04 (1 H, dd, J = 5.4, 8.3 Hz), 4.43 (1 H, m), 4.18 (1 H, m), 3.79 (6 H, s), 3.49 (1H, AB dd, J = 2.7, 10.8 Hz), 3.40 (1H, AB dd, J = 2.7, 10.6 Hz), 1.65 (9 H, s) ppm; APT (MeOD, 75 MHz) δ 28.0 (CH3), 49.9 (C) 55.8 (CH3), 57.3 (CH), 65.1 (CH2), 72.6 (CH), 87.4 (CH), 87.8 (CH), 87.8 (C), 88.5 (C), 103.0 (CH), 114.4 (CH), 120.1 (CH), 121.8 (CH), 128.2 (CH), 129.1, 129.5 (CH), 131.4 (CH), 132.7 (CH), 136.6 (C), 136.9 (C), 140.1 (C), 142.5 (CH), 145.8 (C), 147.8 (C), 152.6 (C), 160.3 (C), 165.9 (C), 168.9 (C) ppm; IR (KBr plate) ν 3306, 2929, 1766, 1722, 1644, 1614, 1462, 1384, 1254, 1091, 988, 836 cm-1; ESI-MS (positive mode) 303.3 (DMTr) +, 665.6 (M-Boc) +, 766.0 (M) +.
2’-[3-(imidazol-4-yl]acryloyl]-amido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (6)
A solution of urocanic acid (1, 152 mg, 1.1 mmol) in DMF (5 mL) was cooled to 0 °C. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (210 mg, 1.1 mmol) and N-hydroxy-succinimide (127 mg, 1.1 mmol) were added. The mixture was allowed to warm to rt and was stirred for 5h, then 4 (600 mg, 1.1 mmol) was added and the reaction was stirred overnight at rt. H2O was added and the aqueous layer was extracted with EtOAc (3 x) and CHCl3 (1 x). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH, 9:1) to afford 6 (338 mg, 45%): Rf (DCM/ MeOH 9/ 1): 0.2; 1H-NMR (300 MHz, MeOD) δ 7.93 (1 H, d, J = 8.1 Hz), 7.77 (1 H, s), 7.51-7.23 (11 H, m), 6.91 (4 H, d, J = 8.8 Hz), 6.62 (1 H, d, J = 15.6 Hz), 6.17 (1 H, d, J= 8.3 Hz), 5.34 (1 H, d, J = 8.1 Hz), 5.03 (1 H, dd, J = 5.5, 8.3 Hz), 4.42 (1 H, m), 4.18 (1 H, m), 3.79 (6 H, s), 3.49 (1 H, AB dd, J = 2.7, 10.8 Hz), 3.40 (1 H, AB dd, J = 2.7, 10.7 Hz) ppm; APT (MeOD, 75 MHz) δ 169.5 (C), 165.9 5 (C), 160.4 (C), 152.6 (C), 145.8 (C), 142.5 (CH), 136.9 (C), 136.6 (C), 131.5 (CH), 131.4 (CH), 129.5 (CH), 129.0 (CH), 128.1 (CH), 118.7 (CH), 114.3 (CH), 103.0 (CH), 88.5 (C), 87.9 (CH), 87.4 (CH), 72.6 (CH), 65.1 (CH2), 57.2 (CH), 55.8 (CH3) ppm; IR (KBr plate) ν 3252, 2925, 1760, 1652, 1614, 1514, 1463, 1384, 1254, 1178, 1014, 836 cm-1; ESI-MS (positive mode): 303.0 (DMTr) +, 665.8 (M) +.
4-allyloxy-4-oxo-butanoic acid (8)
To a solution of succinic anhydride (7, 2 g 2.0 mmol) in toluene (45 mL) was added 4-dimethyl-aminopyridine (a catalytic amount). Allyl alcohol (1.36 mL, 2.0 mmol) was added dropwise and the reaction mixture was refluxed for 5h. The solvent was removed under reduced pressure and the residue was purified by bulb-to-bulb distillation (0.04 mmHg, 140 °C) to afford 8 (quantitative): 1H-NMR (300 MHz, CDCl3) δ 5.89 (1 H, dddd, J = 5.6, 5.6, 11.1, 16.8 Hz), 5.30 (1 H, dd, J = 1.4, 17.2 Hz), 5.22 (1 H, dd, J = 1.0, 10.4 Hz), 4.58 (2 H, d, J = 5.7 Hz), 2.65 (4 H, t, J = 3.4 Hz) ppm; APT (75 MHz, DMSO) δ 173.3 (C), 171.7 (C), 132.6 (CH), 117.5 (CH2), 64.4 (CH2), 28.6 (CH2), 28.5 (CH2) ppm; IR (KBr film) ν 3090, 2940, 1737, 1650, 1415, 1346, 1170, 992, 936, 842, 558 cm-1; ESI-MS (negative mode) 158.6 (M) +.
2’-[(4-allyloxy-4-oxo)-butanoyl]-amido-2’-deoxyuridine (10)
A solution of 8 (182 mg, 1.15 mmol) in DMF (3 mL) was cooled to 0 °C. N,N’-Dicyclohexyl-carbodiimide (285 mg, 1.38 mmol) and 1-hydroxybenzotriazole (187 mg, 1.38 mmol) were added. The mixture was stirred at 0 °C for 30 min and 9 (280 mg, 1.15 mmol) was added. The mixture was allowed to warm up to rt and stirred overnight. The reaction mixture was filtered to remove the precipitated DCU and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (gradient elution DCM/MeOH, 95:5 to 9:1) to afford 10 (347 mg, 75%): Rf (DCM/MeOH, 8:2): 0.46; 1H-NMR (500 MHz, MeOD) δ 8.00 (1 H, d, J = 8.1 Hz), 6.06 (1 H, d, J = 8.6 Hz), 5.92 (1 H, m), 5.74 (1 H, d, J = 8.1 Hz), 5.30 (1 H, dd, J = 1.5, 17.1 Hz), 5.21 (1 H, dd, J = 1.3, 10.5 Hz), 4.61 (2 H, dd, J = 5.6, 8.5 Hz), 4.56 (1 H, dt, J = 1.3, 5.5 Hz), 4.26 (1 H, dd, J = 1.2, 5.6 Hz), 4.09 (1 H, m), 3.77 (1 H, AB dd, J = 3.1, 12.1 Hz), 3.73 (1 H, AB dd, J = 3.1, 12.1 Hz), 2.59 (4 H, m) ppm; APT (75 MHz, MeOD) 175.0 (C), 174.1 (C), 166.1 (C), 152.8 (C), 142.7 (CH), 133.7 (CH), 118.3 (CH2), 103.3 (CH), 88.6 (CH), 88.1 (CH), 72.2 (CH), 66.4 (CH2), 62.1 (CH2), 56.9 (CH), 34.8 (CH2), 26.8 (CH2) ppm; IR (KBr plate) ν 3322, 2928, 2850, 2738, 2676, 2481, 2355, 1682, 1626, 1575, 1435, 1398, 1244, 1088 cm-1; ESI-MS (positive mode) 406.0 (M+ Na) +.
2’-[(4-allyloxy-4-oxo)-butanoyl]-amido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (11)
Compound 10 was coevaporated with pyridine (3x) and dried under vacuum overnight. To a solution of 10 (300 mg, 0.78 mmol) in pyridine (1.5 mL) was added 4,4’-dimethoxytritylchloride (397 mg, 1.17 mmol) and the reaction was stirred at rt for 6h. MeOH (100 μL) was added to the cooled reaction at 0 °C and stirred for 10 min. The reaction mixture was evaporated under reduced pressure and EtOAc (10 mL) was added. The organic layer was washed with water (10 mL), saturated NaHCO3 (10 mL) and brine (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (gradient elution: DCM+ 1% Et3N → DCM + 1% Et3N+ 1% MeOH → DCM + 1% Et3N + 3% MeOH) to afford 11 (250 mg, 78%): Rf (DCM/MeOH, 95:5): 0.31, 1H-NMR (500 MHz, MeOD) δ 7.80 (1 H, d, J = 8.1 Hz), 7.41 (2 H, d, J = 7.4 Hz), 7.31-7.20 (7 H, m), 6.87 (4 H, d, J = 8.3 Hz), 6.05 (1 H, d, J = 8.4 Hz), 5.89 (1 H, m), 5.28 (1 H, d, J = 8.2 Hz), 5.28 (1 H, dd, J = 1.6, 17.0 Hz), 5.16 (1 H, dd, J = 1.3, 10.5 Hz), 4.55 (1 H, m), 4.31 (1 H, d, J = 5.5 Hz), 4.11 (1 H, m), 3.76 (6 H, s), 3.47-3.43 (2 H, m), 3.42 (1 H, AB dd, J = 2.9, 10.7 Hz), 3.35 (1H, AB dd, J = 2.6, 10.6 Hz), 2.65-2.54 (4 H, m) ppm, APT (75 MHz, MeOD) 175.0 (C), 174.0 (C), 166.0 (C), 160.4 (C), 152.6 (C), 145.8 (C), 142.4 (CH), 136.9 (C), 136.6 (C), 133.7 (CH), 131.4 (CH), 129.5 (CH), 129.0 (CH), 128.1 (CH), 118.2 (CH2), 114.3 (CH), 88.5 (C), 87.6 (CH), 87.3 (CH), 72.4 (CH), 66.3 (CH2), 65.1 (CH2), 57.1 (CH), 55.8 (CH3), 31.3 (CH2), 30.3 (CH2) ppm; IR (KBr plate) ν 3254, 3055, 2935, 1694, 1607, 1510, 1463, 1383, 1251, 1177, 1091, 1032, 990, 830, 703 cm-1; ESI-MS (positive mode) 303.1 (DMTr) +, 707.9 (M + Na) +.
3-Acetoxy-propanoic acid (13)
Sulfuric acid (5 μL) was added to a solution of β-propiolactone (12, 3 mL, 3.44 g, 47.7 mmol) in glacial acetic acid (14 mL). The mixture was stirred at rt for 4h and the solvent was removed under reduced pressure. The residue was purified by a bulb-to-bulb distillation (0.4 mmHg, 200 °C) to afford 13 (quantitative). 1H-NMR (300 MHz, CDCl3) δ 4.31 (2 H, t, J = 4.31 Hz), 2.67 (2 H, t, J = 6.2 Hz), 2.02 (3 H, s) ppm; APT (CDCl3, 75 MHz) δ 176.6 (C), 170.6 (C), 59.8 (CH2), 33.7 (CH2), 20.8 (CH3) ppm; IR (KBr film) ν 3106, 2672, 1744, 1636, 1618, 1429, 1387, 1368, 1240, 1187, 1043, 988, 820, 607 cm-1; MS (EI) m/z (%) 43 (100), 55 (22), 73 (24), 89 (17), 115 (19), 133 (M+, 6).
2’-(3-acetoxypropanoyl)-amido-2’-deoxyuridine (14)
A solution of 13 (170 mg, 1.3 mmol) in DMF (5 mL) was cooled to 0 °C. N,N’-Dicyclohexyl-carbodiimide (265 mg, 1.3 mmol) and 1-hydroxybenzotriazole (166.5 mg, 1.25 mmol) were added. The mixture was stirred at 0 °C for 1h and 9 (250 mg, 1.05 mmol) was added. The mixture was stirred overnight at rt, filtered to remove the precipitated DCU and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH, 9:1) to afford 14 (175 mg, 48%): Rf (DCM/MeOH, 9:1): 0.21; 1H-NMR (300 MHz, MeOD) δ 7.92 (1 H, d, J = 8.1 Hz), 5.96 (1 H, d, J = 8.4 Hz), 5.64 (1 H, d, J = 8.1 Hz), 4.59 (1 H, dd, J = 5.6, 8.4 Hz), 4.16 (3 H, m), 3.97 (1 H, m), 3.69 (1 H, AB dd, J = 2.8, 12.0 Hz), 3.64 (1 H, AB dd, J = 2.8, 12.0 Hz), 2.47 (2 H, m), 1.90 (3 H, s) ppm; APT (75 MHz, MeOD) 173.5 (C), 172.9 (C), 166.1 (C), 152.7 (C), 142.6 (CH), 103.3 (CH), 88.7 (CH), 88.1 (CH), 72.2 (CH), 63.2 (CH2), 61.4 (CH2), 57.0 (CH), 36.0 (CH2), 20.8 (CH3) ppm; IR (KBr plate) ν 3444, 1667, 1634, 1557, 1463, 1384 cm-1; MS (EI) m/z (%) 43 (100), 52 (47), 77 (30), 107 (23), 157 (4), 314 (M+- Ac, 1), 359 (M+,1).
2’-(3-acetoxypropanoyl)-amido-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyuridine (15)
Compound 14 was coevaporated three times with pyridine and dried under vacuum for 6h. To a solution of 14 (175 mg, 0.378 mmol) in pyridine (2 mL) was added 4,4’-dimethoxytritylchloride (192 mg, 0.567 mmol) and the reaction was stirred overnight at rt. MeOH (100 μL) was added to the cooled reaction mixture at 0 °C and stirred for 10 min. EtOAc (10 mL) was added and the organic layer was washed successively with water (10 mL), saturated NaHCO3 (10 mL) and brine (10 mL). The combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/MeOH, 95:5) to afford 15 (250 mg, 78%): Rf (DCM/MeOH, 9:1): 0.57; 1H-NMR (500 MHz, MeOD) δ 7.83 (1H, d, J = 8.0 Hz), 7.42 (2 H, d, J = 7.6 Hz), 7.31-7.29 (6 H, m), 7.22 (1 H, m), 6.87 (4 H, d, J = 8.6 Hz), 6.06 (1 H, d, J = 8.3 Hz), 5.28 (1 H, d, J = 8.1), 4.86 (1 H, dd, J= 5.5, 8.3 Hz), 4.32 (1 H, m), 4.28 (2 H, m), 4.11 (1 H, m), 3.76 (6 H, s), 3.43 (1 H, AB dd, J = 2.8, 10.6 Hz), 3.36 (1 H, AB dd, J = 2.9, 10.6 Hz), 2.63-2.53 (2 H, m), 1.99 (3 H, s) ppm; APT (75 MHz, MeOD) 173.6 (C), 172.7 (C), 165.9 (C), 160.3 (C), 152.6 (C), 145.8 (C), 142.4 (CH), 136.8 (C), 131.4 (CH), 129.5 (CH), 129.0 (CH), 128.1 (CH), 114.3 (CH), 102.9 (CH), 88.5 (C), 87.6 (CH), 87.4 (CH), 72.4 (CH), 65.1 (CH2), 61.7 (CH2), 57.1 (CH), 55.8 (CH3), 36.1 (CH2), 20.8 (CH3) ppm, IR (KBr plate) ν 3328, 3064, 2931, 2836, 1694, 1607, 1506, 1463, 1385, 1252, 1177, 1092, 1034, 986, 829, 757, 703, 583, 555 cm-1; ESI-MS (positive mode) 303.3 (DMTr) +, 676.8 (M + NH4) +.
Acknowledgements
Financial support from Ghent University (BOF 01115203) and FWO Vlaanderen (KAN 1.5.186.03) is gratefully acknowledged. M.C. would like to thank Ghent University for an assistant position.
References
- Matthews, B. W.; Sigler, P. B.; R., H.; Blow, D. M. Three-dimensional structure of tosyl-chymotrypsin. Nature 1967, 214, 652–656. [Google Scholar] [CrossRef]
- Blow, D. M.; Birktoft, J. J.; Hartley, B. S. Role of a buried acid group in the mechanism of action of chymotryspin. Nature 1969, 221, 337–340. [Google Scholar] [CrossRef]
- Steitz, T. A.; Henderson, R.; Blow, D. M. Structure of crystalline chymotrypsin. J. Mol. Biol. 1969, 46, 337–348. [Google Scholar]
- Blow, D. M. Structure and Mechanism of Chymotrypsin. Acc. Chem. Res. 1976, 9, 145–152. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4523. [Google Scholar] [CrossRef]
- Hahn, K. W.; Klis, W. A.; Stewart, J. M. Design and Synthesis of a Peptide Having Chymotrypsin-Like Esterase-Activity. Science 1990, 248, 1544–1547. [Google Scholar]
- Atassi, M. Z.; Manshouri, T. Design of Peptide Enzymes (Pepzymes) - Surface-Simulation Synthetic Peptides That Mimic the Chymotrypsin and Trypsin Active-Sites Exhibit the Activity and Specificity of the Respective Enzyme. Proc. Nat. Acad. Sci. USA 1993, 90, 8282–8286. [Google Scholar] [CrossRef]
- Corey, M. J.; Hallakova, E.; Pugh, K.; Stewart, J. M. Studies on Chymotrypsin-Like Catalysis by Synthetic Peptides. Appl. Biochem. Biotechnol. 1994, 47, 199–212. [Google Scholar] [CrossRef]
- Corey, D. R.; Phillips, M. A. Cyclic-Peptides as Proteases - a Reevaluation. Proc. Nat. Acad. Sci. USA 1994, 91, 4106–4109. [Google Scholar] [CrossRef]
- Wells, J. A.; Fairbrother, W. J.; Otlewski, J.; Laskowski, M.; Burnier, J. A Reinvestigation of a Synthetic Peptide (Trpepz) Designed to Mimic Trypsin. Proc. Nat. Acad. Sci. USA 1994, 91, 4110–4114. [Google Scholar]
- Walse, B.; Ullner, M.; Lindbladh, C.; Bulow, L.; Drakenberg, T.; Teleman, O. Structure of a cyclic peptide with a catalytic triad, determined by computer simulation and NMR spectroscopy. J. Comput.-Aided Mol. Des. 1996, 10, 11–22. [Google Scholar] [CrossRef]
- Fukushima, Y. Enantioselectivity enhancement of ester cleavage by a beta-sheet polypeptide containing catalytic triads in a serine protease. B. Chem. Soc. Jpn. 1996, 69, 2269–2274. [Google Scholar] [CrossRef]
- Stavrakoudis, A.; Demetropoulos, I.; Sakarellos, C.; Sakarellos-Daaitsiotis, M.; Tsikaris, V. Design, synthesis and catalytic activity of a serine protease synthetic model. Lett. Pept. Sci. 1997, 4, 481–487. [Google Scholar]
- Broo, K. S.; Brive, L.; Ahlberg, P.; Baltzer, L. Catalysis of hydrolysis and transesterification reactions of p-nitrophenyl esters by a designed helix-loop-helix dimer. J. Am. Chem. Soc. 1997, 119, 11362–11372. [Google Scholar]
- Li, Y. S.; Zhao, Y. F.; Hatfield, S.; Wan, R.; Zhu, Q.; Li, X. H.; McMills, M.; Ma, Y.; Li, J.; Brown, K. L.; He, C.; Liu, F.; Chen, X. Z. Dipeptide seryl-histidine and related oligopeptides cleave DNA, protein, and a carboxyl ester. Bioorg. Med. Chem. 2000, 8, 2675–2680. [Google Scholar] [CrossRef]
- Delort, E.; Darbre, T.; Reymond, J. A strong positive dendritic effect in a peptide dendrimer-catalyzed ester hydrolysis reaction. J. Am. Chem. Soc. 2004, 126, 15642–15643. [Google Scholar] [CrossRef]
- Dsouza, V. T.; Bender, M. L. Miniature Organic-Models of Enzymes. Accounts Chem. Res. 1987, 20, 146–152. [Google Scholar] [CrossRef]
- Zimmerman, S. C. On the Evaluation of a Small Molecule Mimic of Chymotrypsin. Tetrahedron Lett. 1989, 30, 4357–4358. [Google Scholar]
- Rao, K. R.; Srinivasan, T. N.; Bhanumathi, N.; Sattur, P. B. Artificial Enzymes - Synthesis of Imidazole Substituted at C(2) of Beta-Cyclodextrin as an Efficient Enzyme Model of Chymotrypsin. J. Chem. Soc. Chem. Comm. 1990, 10–11. [Google Scholar]
- Breslow, R. Biomimetic Chemistry and Artificial Enzymes - Catalysis by Design. Acc. Chem. Res. 1995, 28, 146–153. [Google Scholar] [CrossRef]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–27. [Google Scholar] [CrossRef]
- Madder, A.; De Clercq, P.; Declercq, J.P. Stepwise Approach toward First Generation Nonenzymatic Hydrolases. J. Org. Chem. 1998, 63, 2548–2559. [Google Scholar] [CrossRef]
- De Muynck, H.; Madder, A.; Farcy, N.; De Clercq, P.; Pérez-Payan, M.; Öhberg, L. ; Davis, A. Application of Combinatorial Procedures in the Search for Serine-Protease-Like Activity with Focus on the Acyl Transfer Step. Angew. Chem. Int. Ed. 2000, 39, 145–148. [Google Scholar] [CrossRef]
- Madder, A.; Li, L.; De Muynck, H.; Farcy, N; Van Haver, D.; Fant, F.; Vanhoenacker, G.; Sandra, P.; Davis, A.; De Clercq, P. Evaluation of a Two-Stage Screening Procedure in the Combinatorial Search for Serine Protease-Like Activity. J. Comb. Chem. 2002, 4, 552–562. [Google Scholar] [CrossRef]
- Gea, A.; Farcy, N.; Roqué i Rossell, N.; Martins, J.; De Clercq, P.; Madder, A. Solid-Supported Synthesis of Highly Functionalized Tripodal Peptides with flexible but Preorganized Geometry: Towards Potential Serine Protease Mimics. Eur. J. Org. Chem. 2006, 4135–4146. [Google Scholar]
- Beaucage, S. L.; Caruthers, M. H. Deoxynucleoside Phosphoramidites - a New Class of Key Intermediates for Deoxypolynucleotide Synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- Verheyden, J. P.; Wagner, D.; Moffatt, J. G. Synthesis of some pyrimidine 2'-amino-2'-deoxynucleosides. J. Org. Chem. 1971, 36, 250–254. [Google Scholar] [CrossRef]
- Kirschenheuter, G. P.; Zhai, Y.; Pieken, W. A. An improved synthesis of 2'-azido-2'deoxyuridine. Tetrahedron Lett. 1994, 35, 8517–8520. [Google Scholar] [CrossRef]
- Kohgo, S.; Shinozuka, K.; Ozaki, H.; Sawai, H. Synthesis of a novel 2 '-deoxyuridine derivative bearing a cyanomethoxycarbonylmethyl group at C-5 position and its use for versatile post-synthetic functionalization of oligodeoxyribonucleotides. Tetrahedron Lett. 1998, 39, 4067–4070. [Google Scholar] [CrossRef]
- Beloglazova, N. G.; Fabani, M. M.; Zenkova, M. A.; Bichenkova, E. V.; Polushin, N. N.; Sil'nikov, V. V.; Douglas, K. T.; Vlassov, V. V. Sequence-specific artificial ribonucleases. I. Bis-imidazole-containing oligonucleotide conjugates prepared using precursor-based strategy. Nucleic Acids Res. 2004, 32, 3887–3897. [Google Scholar] [CrossRef]
- Beban, M.; Miller, P. S. Preparation of an imidazole-conjugated oligonucleotide. Bioconjugate Chem. 2000, 11, 599–603. [Google Scholar] [CrossRef]
- Wang, G. Y.; Bergstrom, D. E. Synthesis of Oligonucleotides Containing N2-[2-(Imidazol-4-Ylacetamido)Ethyl]-2'-Deoxyguanosine. Tetrahedron Lett. 1993, 34, 6725–6728. [Google Scholar]
- Heeb, N. V.; Benner, S. A. Guanosine Derivatives Bearing an N-2-3-Imidazolepropionic Acid. Tetrahedron Lett. 1994, 35, 3045–3048. [Google Scholar] [CrossRef]
- Truffert, J. C.; Asseline, U.; Brack, A.; Thuong, N. T. Synthesis, purification and characterization of two peptide-oligonucleotide conjugates as potential artificial nucleases. Tetrahedron 1996, 52(8), 3005–3016. [Google Scholar] [CrossRef]
- Holmes, S. C.; Gait, M. J. Syntheses and oligonucleotide incorporation of nucleoside analogues containing pendant imidazolyl or amino functionalities - The search for sequence-specific artificial ribonucleases. Eur. J. Org. Chem. 2005, 5171–5183. [Google Scholar] [CrossRef]
- Bashkin, J. K.; Gard, J. K.; Modak, A. S. Synthesis and Characterization of Nucleoside Peptides - toward Chemical Ribonucleases .1. J. Org. Chem. 1990, 55, 5125–5132. [Google Scholar] [CrossRef]
- Bashkin, J. K.; Mcbeath, R. J.; Modak, A. S.; Sample, K. R.; Wise, W. B. Synthesis and Characterization of Oligonucleotide Peptides. J. Org. Chem. 1991, 56, 3168–3176. [Google Scholar] [CrossRef]
- Zaramella, S.; Stromberg, R.; Yeheskiely, E. Stability studies of N-acylimidazoles. Eur. J. Org. Chem. 2002, 2633–2639. [Google Scholar] [CrossRef]
- Verbeure, B.; Lacey, C. J.; Froeyen, M.; Rozenski, J.; Herdewijn, P. Synthesis and cleavage experiments of oligonucleotide conjugates with a diimidazole-derived catalytic center. Bioconj. Chem. 2002, 13, 333–350. [Google Scholar]
- Lermer, L.; Roupioz, Y.; Ting, R.; Perrin, D. M. Toward an RNaseA mimic: a DNAzyme with imidazoles and cationic amines. J. Am. Chem. Soc. 2002, 124, 9960–9961. [Google Scholar]
- Santoro, S. W.; Joyce, G. F.; Sakthivel, K.; Gramatikova, S.; Barbas, C. F. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000, 122, 2433–2439. [Google Scholar]
- Lapidot, Y.; Rappoport, S.; Wolman, Y. Use of esters of N-hydroxysuccinimide in the synthesis of N-acylamino acids. J. Lipid Res. 1967, 8, 142–145. [Google Scholar]
- Purwanto, M. G. M.; Lengeler, D.; Weisz, K. Nucleosides derived from urocanic acid: potential ligands for CG base pairs. Tetrahedron Lett. 2002, 43, 61–64. [Google Scholar] [CrossRef]
- Winkler, J.; Urban, E.; Losert, D.; Wacheck, V.; Pehamberger, H.; Noe, C. A novel concept for ligand attachment to oligonucleotides via a 2'-succinyl linker. Nucleic Acids Res. 2004, 32, 710–718. [Google Scholar] [CrossRef]
- Berthod, T.; Petillot, Y.; Guy, A.; Cadet, J.; Molko, D. Synthesis of oligonucleotides containing 5-carboxy-2'-deoxyuridine at defined sites. J. Org. Chem. 1996, 61, 6075–6078. [Google Scholar] [CrossRef]
- Guerniou, V.; Gasparutto, D.; Sauvaigo, S.; Favier, A.; Cadet, J. New synthesis of 5-carboxy-2 '-deoxyuridine and its incorporation into synthetic oligonucleotides. Nucleos. Nucleot. Nuc. 2003, 22, 1073–1075. [Google Scholar] [CrossRef]
- Kachalova, A.; Zubin, E.; Stetsenko, D.; Gait, M.; Oretskaya, T. Oligonucleotides with 2 '-O-carboxymethyl group: synthesis and 2 '-conjugation via amide bond formation on solid phase. Org. Biomol. Chem. 2004, 2, 2793–2797. [Google Scholar] [CrossRef]
- Grandas, A.; Marchan, V.; Debéthune, L.; Pedroso, E. Stepwise solid-phase synthesis of nucleopeptides. Curr. Prot. Nucl. Acid Chem. 2004, 4.22.1–4.22.54. [Google Scholar]
- Lioy, E.; Suarez, J.; Guzman, F.; Siegrist, S.; Pluschke, G.; Patarroyo, M. E. Synthesis, biological, and immunological properties of cyclic peptides from Plasmodium falciparum merozoite surface protein-1. Angew. Chem. Int. Ed. 2001, 40, 2631–2635. [Google Scholar] [CrossRef]
- Kawai, K.; Kawabata, K.; Tojo, S.; Majima, T. Synthesis of ODNs containing 4-methylamino-1,8-naphthalimide as a fluorescence probe in DNA. Bioorg. Med. Chem. Lett. 2002, 12, 2363–2366. [Google Scholar] [CrossRef]
- Zatsepin, T. S.; Stetsenko, D. A.; Gait, M. J.; Oretskaya, T. S. Synthesis of DNA conjugates by solid-phase fragment condensation via aldehyde-nucleophile coupling. Tetrahedron Lett. 2005, 46(18), 3191–3195. [Google Scholar] [CrossRef]
- Casimir, J. R.; Turetta, C.; Ettouati, L.; Paris, J. First Application of the Dakin-West Reaction to Fmoc Chemistry - Synthesis of the Ketomethylene Tripeptide Fmoc-N-Alpha-Asp(Tbu)-(R,S)Tyr(Tbu)Psi(Co-Ch2)Gly-Oh. Tetrahedron Lett. 1995, 36, 4797–4800. [Google Scholar]
- Chenna, A.; Singer, B. Large-Scale Synthesis of P-Benzoquinone-2'-Deoxycytidine and P-Benzoquinone-2'-Deoxyadenosine Adducts and Their Site-Specific Incorporation into DNA Oligonucleotides. Chem. Res. Toxicol. 1995, 8, 865–874. [Google Scholar] [CrossRef]
- Chenna, A.; Singer, B. Synthesis of a benzene metabolite adduct, 3''-hydroxy-1,N-2-benzetheno-2'-deoxyguanosine, and its site-specific incorporation into DNA oligonucleotides. Chem. Res. Toxicol. 1997, 10, 165–171. [Google Scholar] [CrossRef]
- Lawrence, A. J.; Pavey, J. B. J.; Cosstick, R.; ONeil, I. A. Synthesis and properties of 2'-deoxy-2'-alpha-C-branched nucleosides and nucleotides. J. Org. Chem. 1996, 61, 9213–9222. [Google Scholar]
- Pavey, J. B. J.; Cosstick, R.; O'Neill, I. A. The synthesis of a novel 2 ',3 '-cyclic phosphate derived from 2 '-homouridine. Tetrahedron Lett. 1998, 39, 8923–8924. [Google Scholar] [CrossRef]
- Pavey, J. B. J.; Lawrence, A. J.; Potter, A. J.; Cosstick, R.; O'Neil, I. A. The synthesis of 2 '-homouridine, its incorporation into a dinucleoside monophosphate and hydrolytic behaviour of the dimer. Tetrahedron Lett. 1998, 39, 6967–6970. [Google Scholar] [CrossRef]
- Lawrence, A.; Pavey, J.; O’Neil, I.; Cosstick, R. Synthesis of Functionalised 2’-C-Branched Nucleosides via Their γ-Butyrolactones. Tetrahedron Lett. 1995, 36, 6341–6344. [Google Scholar] [CrossRef]
- Asseline, U.; Chassignol, M.; Draus, J.; Durand, M.; Maurizot, J. Synthesis and Properties of Oligo-2’-deoxyribonucleotides Containing Internucleotidic Phosphoramidate Linkages Modified with Pendant Groups Ending with either Two Amino or Two Hydroxyl Fucntions. Bioorg. Med. Chem. 2003, 11, 3499–3511. [Google Scholar] [CrossRef]
- Gefflaut, T.; Blonski, C.; Perie, J. Slow reversible inhibitions of rabbit muscle aldolase with substrate analogues: Synthesis, enzymatic kinetics and UV difference spectroscopy studies. Bioorg. Med. Chem. 1996, 4, 2043–2054. [Google Scholar] [CrossRef]
- Sample Availability: No samples available
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Catry, M.A.; Madder, A. Synthesis of Functionalised Nucleosides for Incorporation into Nucleic Acid-Based Serine Protease Mimics. Molecules 2007, 12, 114-129. https://doi.org/10.3390/12010114
AMA Style
Catry MA, Madder A. Synthesis of Functionalised Nucleosides for Incorporation into Nucleic Acid-Based Serine Protease Mimics. Molecules. 2007; 12(1):114-129. https://doi.org/10.3390/12010114
Chicago/Turabian StyleCatry, Mieke A., and Annemieke Madder. 2007. "Synthesis of Functionalised Nucleosides for Incorporation into Nucleic Acid-Based Serine Protease Mimics" Molecules 12, no. 1: 114-129. https://doi.org/10.3390/12010114