A Convenient Approach to Heterocyclic Building Blocks: Synthesis of Novel Ring Systems Containing a [5,6]Pyrano[2,3-c]pyrazol-4(1H)-one Moiety

Abstract
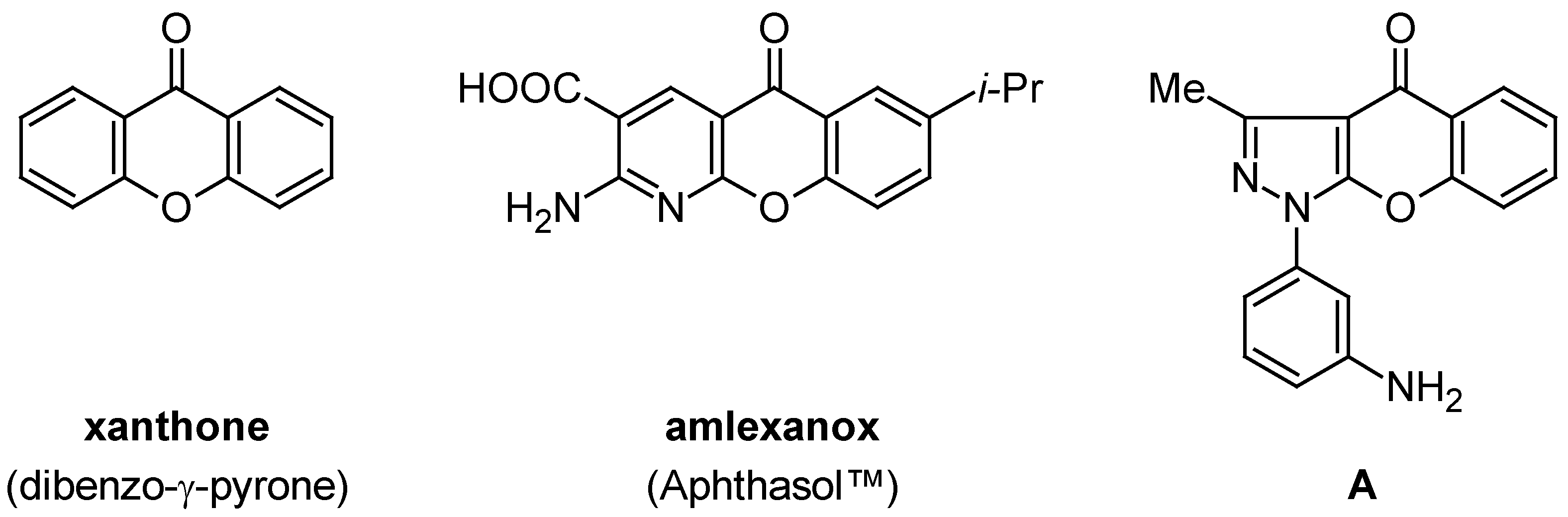
:Introduction

Results and Discussion
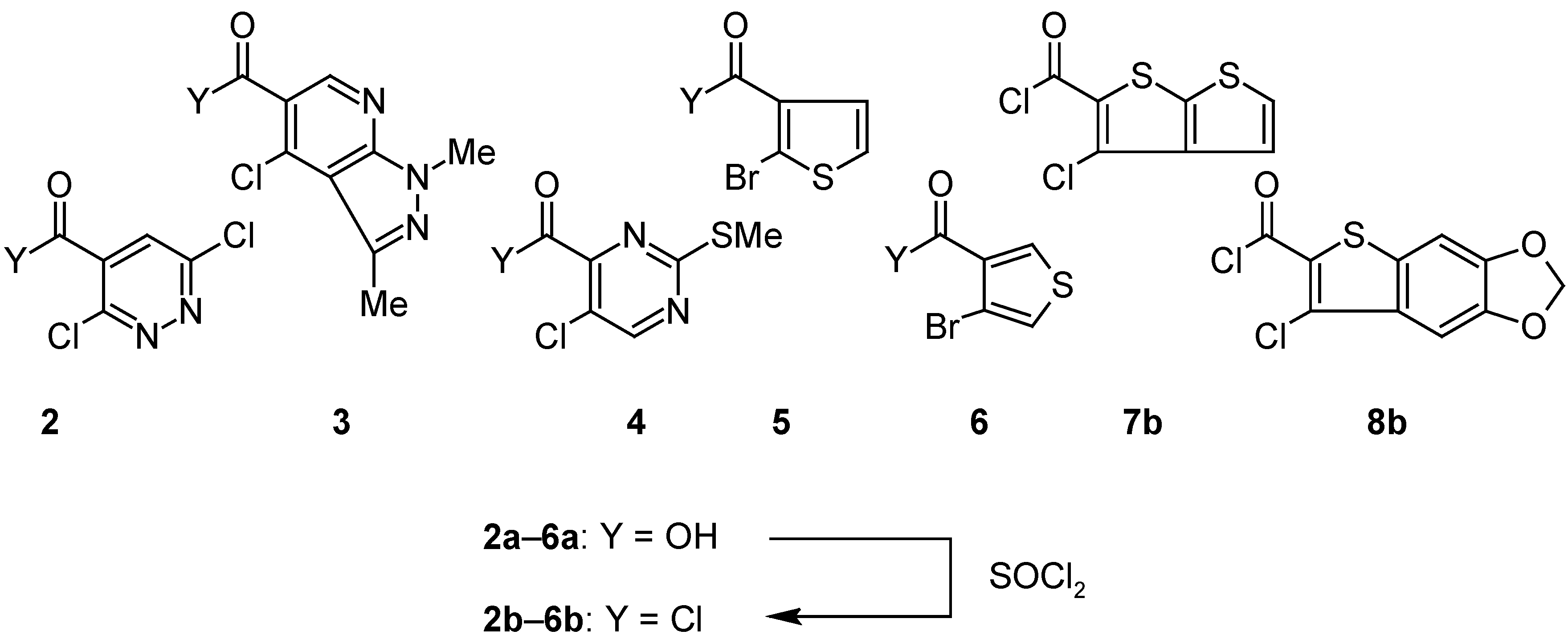
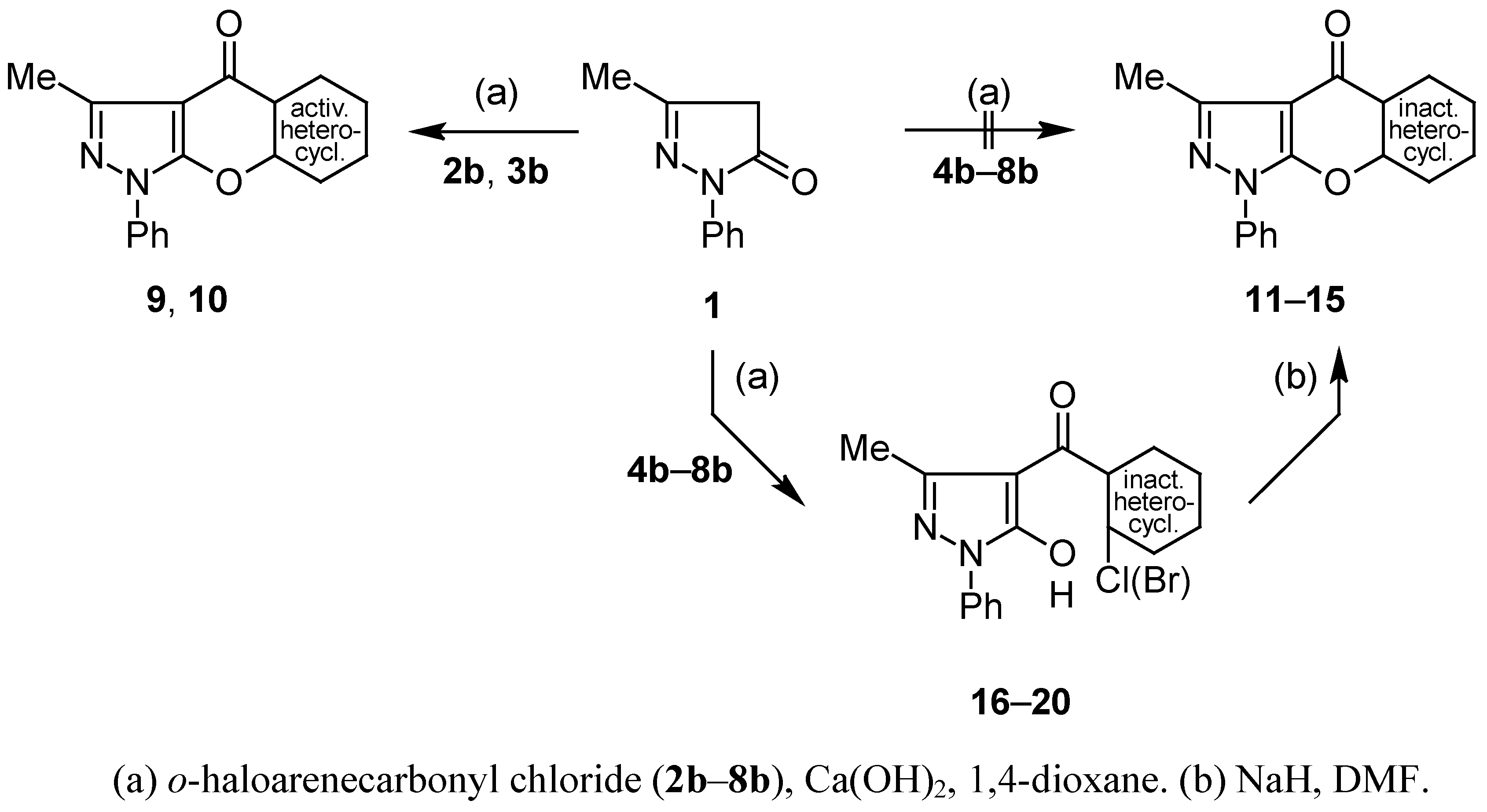
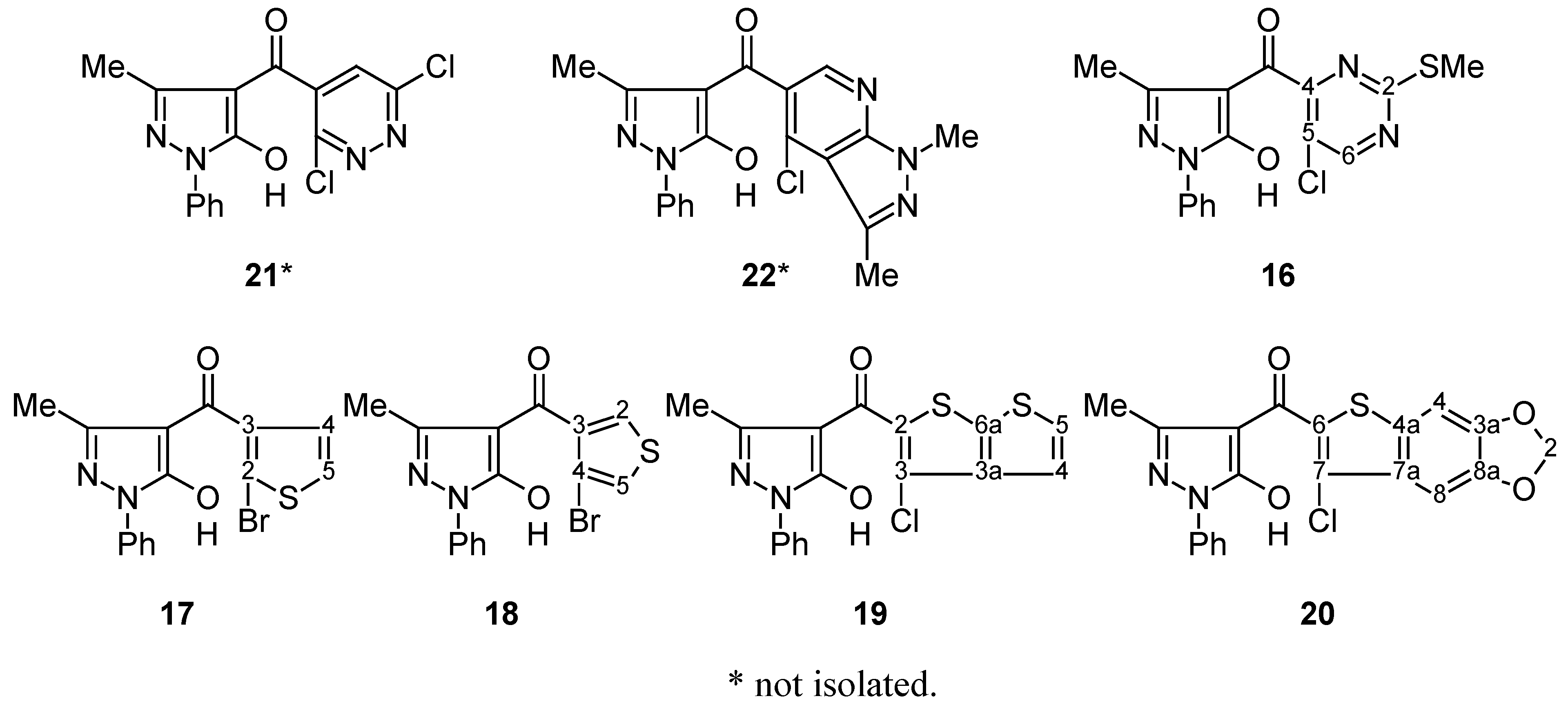
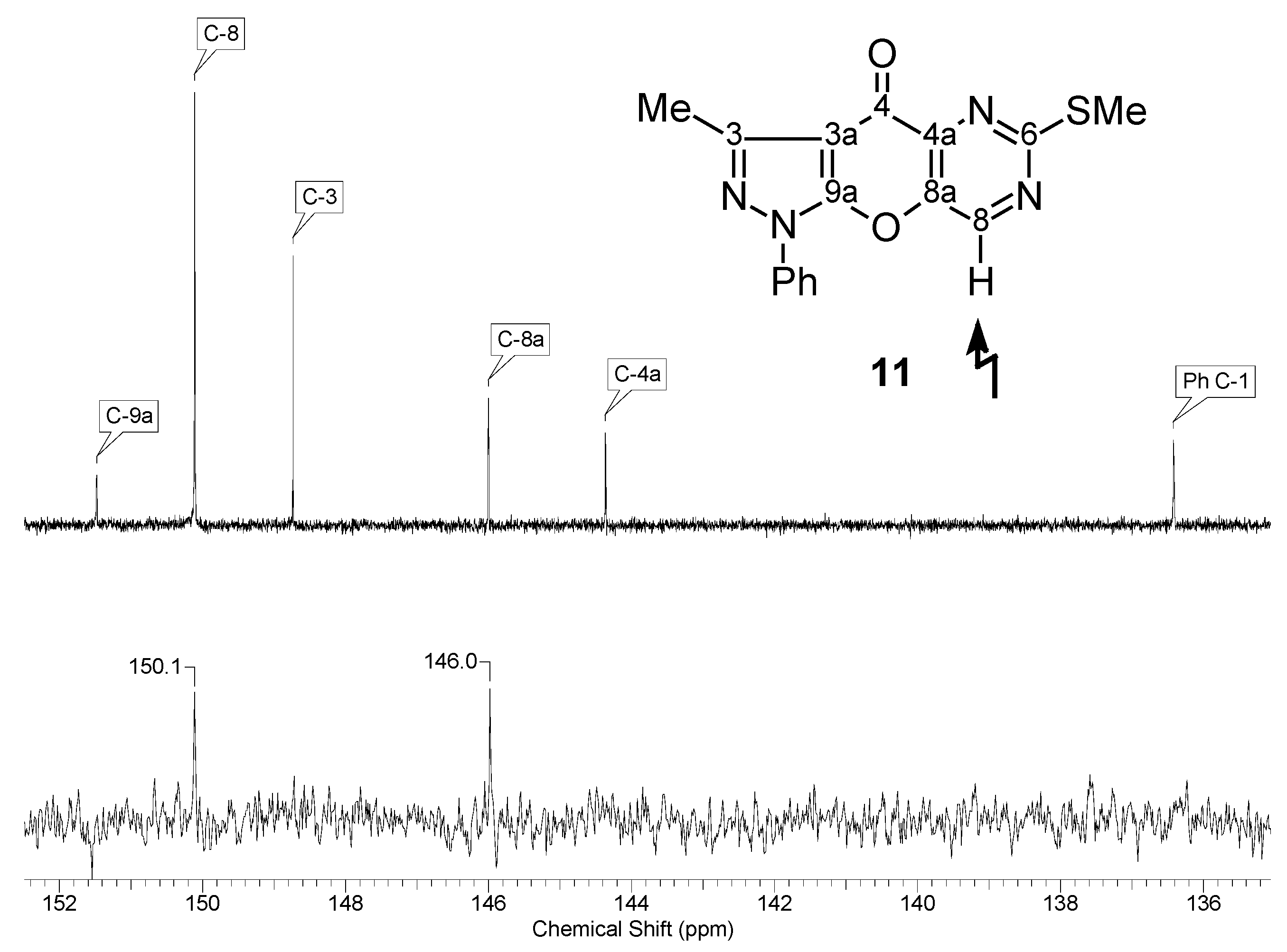
NMR spectroscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | NMR data |
|---|---|---|
| 9 | 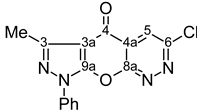 | 1H-NMR (500 MHz, CDCl3) δ: 2.66 (3H, s, Me), 7.43 (1H, m, Ph H-4), 7.56 (2H, m, Ph H-3,5), 7.92 (2H, m, Ph H-2,6), 8.36 (1H, s, H-5). 13C-NMR (125 MHz, CDCl3) δ: 14.0 (Me, 1J=129.7 Hz), 105.1 (C-3a, 3JC3,Me=2.8 Hz), 121.6 (Ph C-2,6), 122.7 (C-4a, 2JC4a,H5=1.3 Hz), 126.9 (C-5, 1J=179.0 Hz), 128.3 (Ph C-4), 129.7 (Ph C-3,5), 136.1 (Ph C-1), 148.7 (C-3, 2JC3,Me=7.2 Hz), 152.2 (C-9a), 155.5 (C-6, 2JC6,H5=0.9 Hz), 160.3 (C-8a, 3JC8a,H5=7.1 Hz), 169.7 (C-4, 3JC4,H5=4.3 Hz). 15N-NMR (50 MHz, CDCl3) δ: −189.5 (N-1), −89.6 (N-2), −8.6 (N-8), 19.9 (N-7). |
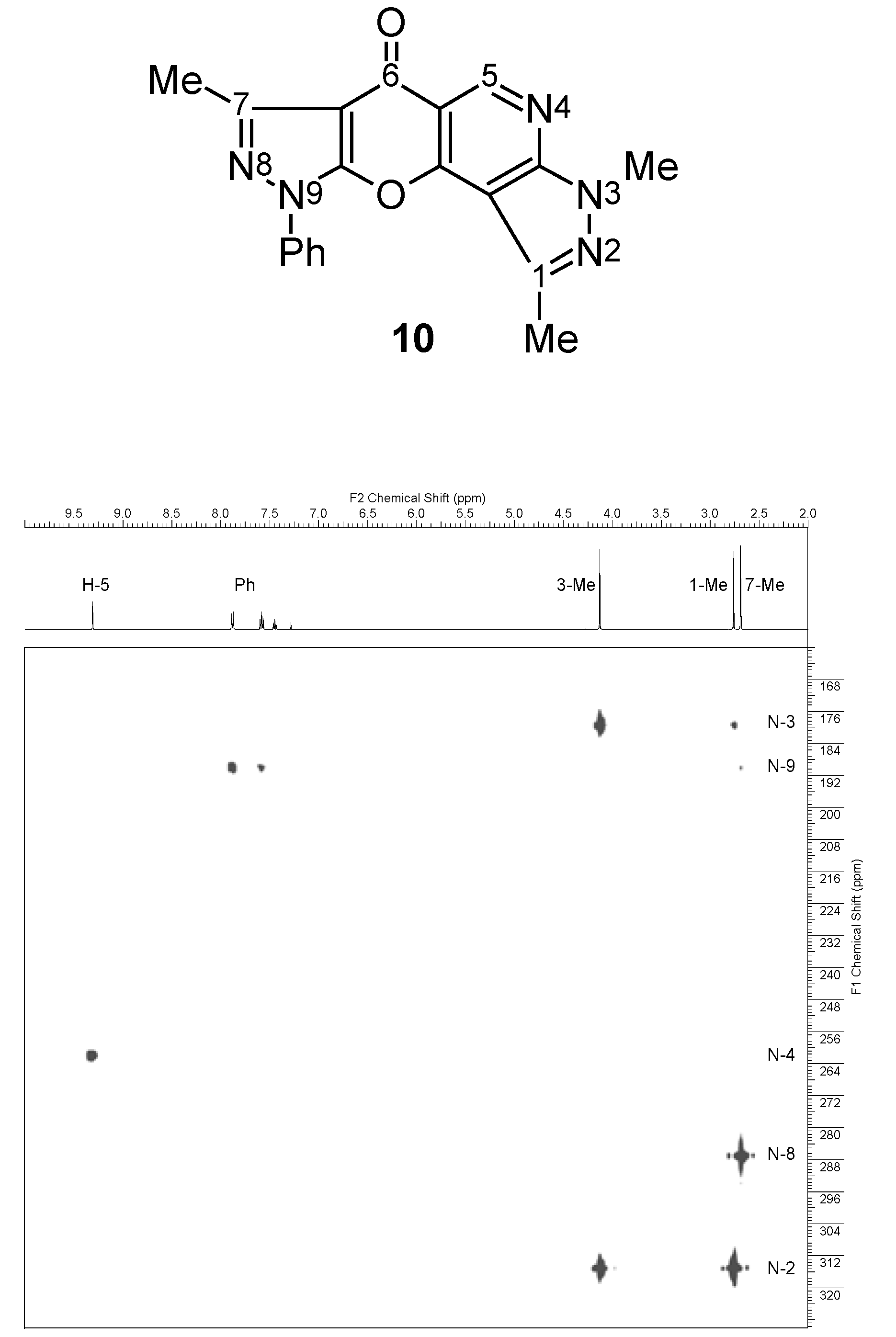
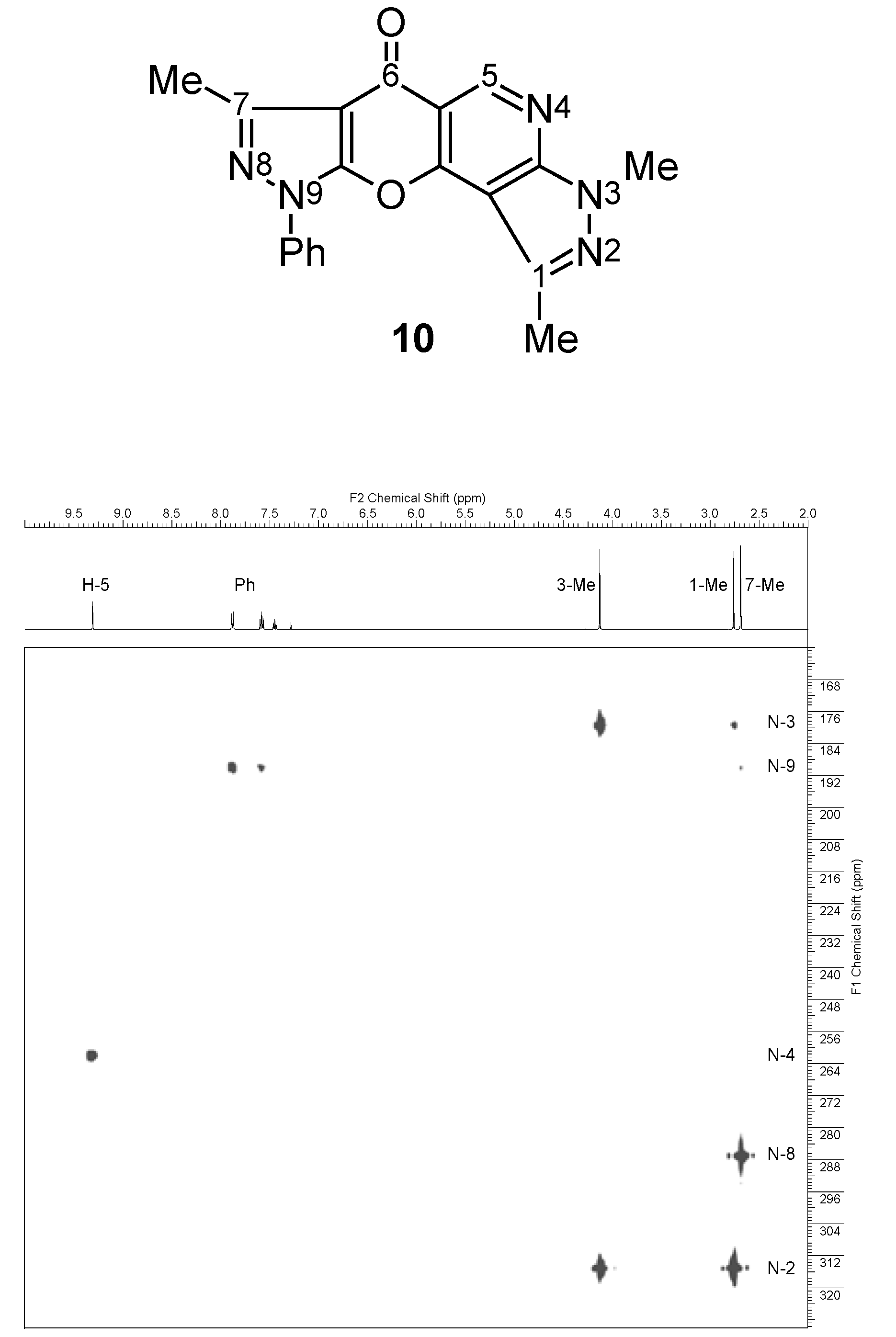
| 10 |  | 1H-NMR (300 MHz, CDCl3) δ: 2.67 (3H, s, 7-Me), 2.74 (3H, s, 1-Me), 4.11 (3H, s, NMe), 7.42 (1H, m, Ph H-4), 7.56 (2H, m, Ph H-3,5), 7.87 (2H, m, Ph H-2,6), 9.30 (1H, s, H-5). 1H-NMR (300 MHz, DMSO-d6) δ: 2.59 (3H, s, 7-Me), 2.68 (3H, s, 1-Me), 4.05 (3H, s, NMe), 7.50 (1H, m, Ph H-4), 7.65 (2H, m, Ph H-3,5), 7.94 (2H, m, Ph H-2,6), 9.17 (1H, s, H-5). 13C-NMR (75 MHz, CDCl3) δ: 14.0 (7-Me, 1J=129.4 Hz), 14.3 (1-Me, 1J=128.8 Hz), 34.1 (NMe, 1J=140.9 Hz), 103.3 (C-10b, 3JC10b,1Me=3.0 Hz, 4JC10b,H5=1.2 Hz), 105.8 (C-6a, 3JC6a,7Me=2.8 Hz), 111.8 (C-5a, 2JC5a,H5=7.9 Hz), 121.3 (Ph C-2,6), 127.9 (Ph C-4), 129.5 (Ph C-3,5), 136.6 (Ph C-1), 140.4 (C-1, 2JC1,1Me=7.2 Hz), 148.3 (C-7, 2JC7,7Me=7.2 Hz), 149.9 (C-5, 1J=186.2 Hz), 151.6 (C-9a), 154.4 (C-3a, 3JC3a,NMe=2.1 Hz, 3JC3a,H5=15.5 Hz), 155.5 (C-10a, 3JC10a,H5=8.3 Hz), 172.6 (C-6, 3JC6,H5=2.2 Hz). 15N-NMR (50 MHz, CDCl3) δ: −202.4 (N-3), −191.6 (N-9), −119.8 (N-4), −94.8 (N-8), −66.8 (N-2). |
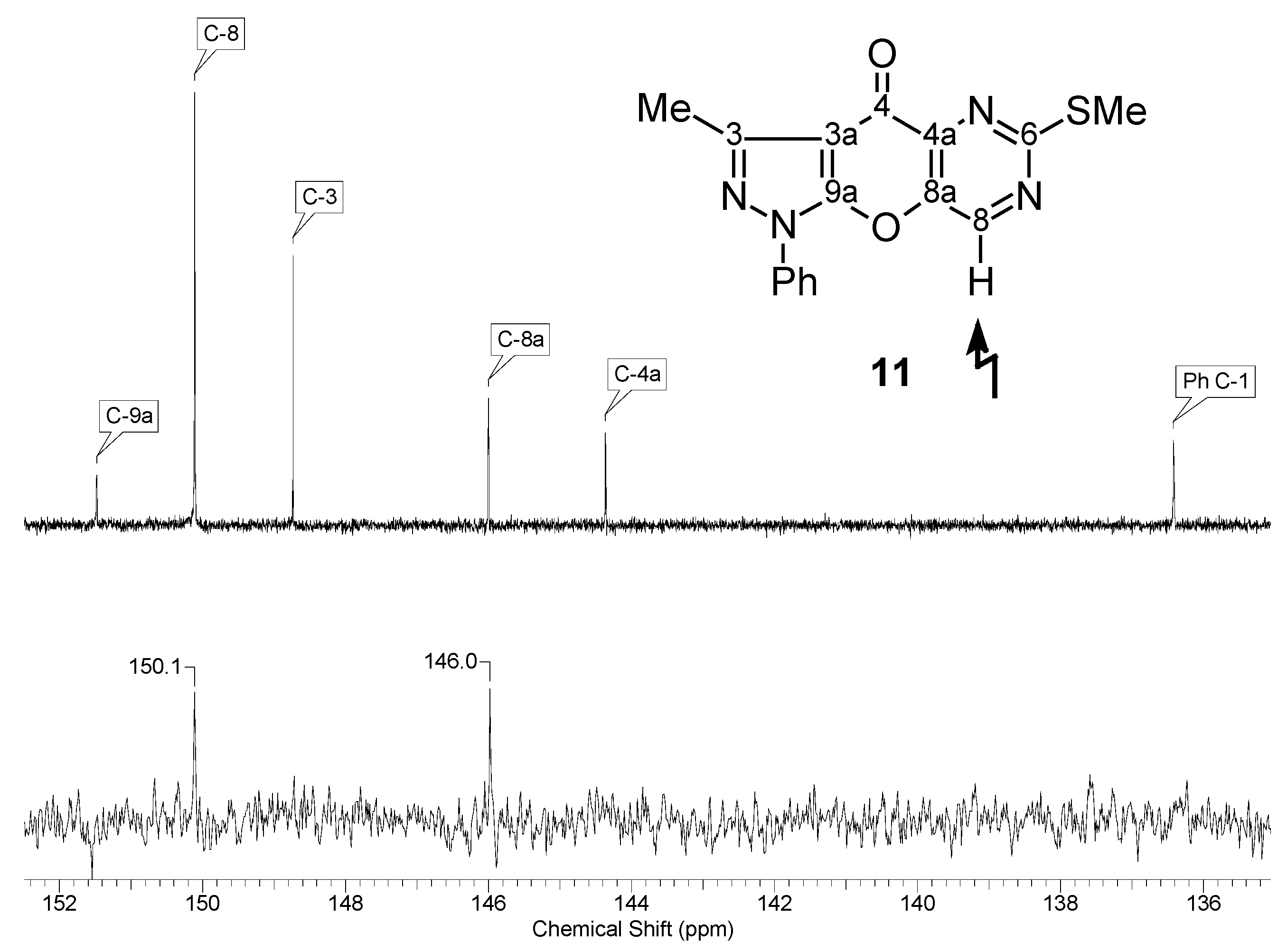
| 11 | 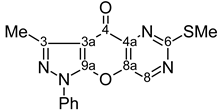 | 1H-NMR (300 MHz, CDCl3) δ: 2.68 (3H, s, 3-Me), 2.68 (3H, s, SMe), 7.42 (1H, m, Ph H-4), 7.55 (2H, m, Ph H-3,5), 7.82 (2H, m, Ph H-2,6), 8.96 (1H, s, H-8). 13C-NMR (75 MHz, CDCl3) δ: 14.1 (3-Me, 1J=129.6 Hz), 14.8 (SMe, 1J=141.8 Hz), 107.8 (C-3a, 3JC3a,3Me=2.8 Hz), 121.5 (Ph C-2,6), 128.0 (Ph C-4), 129.6 (Ph C-3,5), 136.4 (Ph C-1), 144.4 (C-4a, 3JC4a,H8=3.0 Hz), 146.0 (C-8a, 2JC8a,H8=3.0 Hz), 148.8 (C-3, 2JC3,3Me=7.2 Hz), 150.1 (C-8, 1J=187.4 Hz), 151.5 (C-9a), 170.2 (C-6, 3JC6,SMe=4.7 Hz, 3JC6,H8=12.8 Hz), 170.7 (C-4, 4JC4,H8=2.0 Hz). 15N-NMR (50 MHz, CDCl3) δ: −191.9 (N-1), −108.9 (N-5), −91.1 (N-2), −80.2 (N-7). |
| 12 | 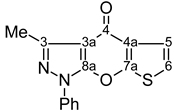 | 1H-NMR (300 MHz, CDCl3) δ: 2.68 (3H, s, Me), 6.95 (1H, d, 3JH6,H5=5.9 Hz, H-6), 7.39 (1H, m, Ph H-4), 7.42 (1H, d, 3JH5,H6=5.9 Hz, H-5), 7.42 (2H, m, Ph H-3,5), 7.81 (2H, m, Ph H-2,6). 13C-NMR (75 MHz, CDCl3) δ: 14.1 (Me, 1J=129.3 Hz), 106.5 (C-3a, 3JC3a,Me=2.7 Hz), 116.2 (C-6, 1J=191.2 Hz, 2JC6,H5=6.5 Hz), 121.3 (Ph C-2,6), 121.4 (C-5, 1J=175.0 Hz, 2JC5,H6=3.5 Hz), 126.0 (C-4a, 2JC4a,H5=4.3 Hz, 3JC4a,H6=8.1 Hz), 127.6 (Ph C-4), 129.5 (Ph C-3,5), 136.8 (Ph C-1), 147.8 (C-3, 2JC3,Me=7.1 Hz), 153.8 (C-8a), 163.7 (C-7a, 3JC7a,H6=8.7 Hz, 3JC7a,H5=11.4 Hz), 171.0 (C-4). 15N-NMR (50 MHz, CDCl3) δ: −193.4 (N-1), −94.8 (N-2). |
| 13 | 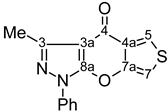 | 1H-NMR (500 MHz, CDCl3) δ: 2.70 (3H, s, Me), 7.15 (1H, d, 3JH7,H5=3.3 Hz, H-7), 7.40 (1H, m, Ph H-4), 7.54 (2H, m, Ph H-3,5), 7.85 (2H, m, Ph H-2,6), 8.12 (1H, d, 3JH5,H7=3.3 Hz, H-5). 13C-NMR (125 MHz, CDCl3) δ: 14.2 (Me, 1J=129.3 Hz), 104.8 (C-3a, 3JC3a,Me=2.5 Hz), 105.7 (C-7, 1J=190.0 Hz, 3JC7,H5=4.3 Hz), 121.6 (Ph C-2,6), 126.3 (C-5, 1J=192.2 Hz, 3JC5,H7=5.9 Hz), 127.5 (Ph C-4), 129.4 (Ph C-3,5), 129.5 (C-4a), 137.0 (Ph C-1), 148.7 (C-3, 2JC3,Me=7.1 Hz), 151.4 (C-7a, 3JC7a,H5=11.2 Hz), 154.7 (C-8a), 171.2 (C-4, 3JC4,H5=2.1 Hz). |
| 14 | 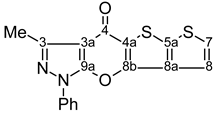 | 1H-NMR (300 MHz, CDCl3) δ: 2.68 (3H, s, Me), 7.40 (1H, d, 3JH8,H7=5.4 Hz, H-8), 7.41 (1H, m, Ph H-4), 7.47 (1H, d, 3JH7,H8=5.4 Hz, H-7), 7.56 (2H, m, Ph H-3,5), 7.87 (2H, m, Ph H-2,6). 13C-NMR (75 MHz, CDCl3) δ: 14.0 (Me, 1J=129.3 Hz), 105.8 (C-3a, 3JC3a,Me=2.8 Hz), 117.9 (C-8, 1J=173.9 Hz, 2JC8,H7=4.1 Hz), 121.2 (Ph C-2,6), 127.0 (C-4a, 5JC4a,H7=0.8 Hz), 127.5 (Ph C-4), 129.5 (Ph C-3,5), 130.2 (C-7, 1J=188.4 Hz, 2JC7,H8=6.9 Hz), 134.9 (C-8a, 2JC8a,H8=5.5 Hz, 3JC8a,H7=10.5 Hz), 137.1 (Ph C-1), 142.6 (C-5a, 3JC5a,H7=7.7 Hz, 3JC5a,H8=9.6 Hz), 147.2 (C-3, 2JC3,Me=7.1 Hz), 147.5 (C-8b, 3JC8b,H8=1.0 Hz, 4JC8b,H7=0.8 Hz), 153.1 (C-9a), 170.2 (C-4). 15N-NMR (50 MHz, CDCl3) δ: −193.2 (N-1), −93.8 (N-2). |
| 15 | 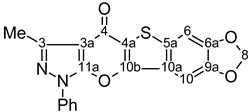 | 1H-NMR (500 MHz, CDCl3) δ: 2.71 (3H, s, Me), 6.13 (2H, s, H-8), 7.24 (1H, s, H-6), 7.34 (1H, s, H-10), 7.43 (1H, m, Ph H-4), 7.59 (2H, m, Ph H-3,5), 7.91 (2H, m, Ph H-2,6). 13C-NMR (125 MHz, CDCl3) δ: 14.1 (Me, 1J=129.3 Hz), 99.6 (C-10, 1J=168.0 Hz, 4JC10,H6=1.3 Hz), 102.3 (C-8, 1J=174.9 Hz), 102.9 (C-6, 1J=168.7 Hz, 4JC6,H10=1.3 Hz), 106.2 (C-3a, 3JC3a,Me=2.7 Hz), 121.2 (Ph C-2,6), 122.4 (C-4a), 122.5 (C-10a, 3JC10a,H6=6.3 Hz), 127.5 (Ph C-4), 129.6 (Ph C-3,5), 134.7 (C-5a, 2JC5a,H6=1.5 Hz, 3JC5a,H10=7.7 Hz), 137.1 (Ph C-1), 147.3 (C-3, 2JC3,Me=7.1 Hz), 147.7 (C-9a, 2JC9a,H10=4.2 Hz, 3JC9a,H8=1.9 Hz, 3JC9a,H6=6.1 Hz), 149.6 (C-10b, 3JC10b,H10=3.2 Hz, 4JC10b,H6=0.9 Hz), 150.3 (C-6a, 2JC6a,H6=4.0 Hz, 3JC6a,H8=1.9 Hz, 3JC6a,H10=6.9 Hz), 153.3 (C-11a), 170.5 (C-4). 15N-NMR (50 MHz, CDCl3) δ: −193.2 (N-1), −94.5 (N-2). |
Experimental
Materials and Methods
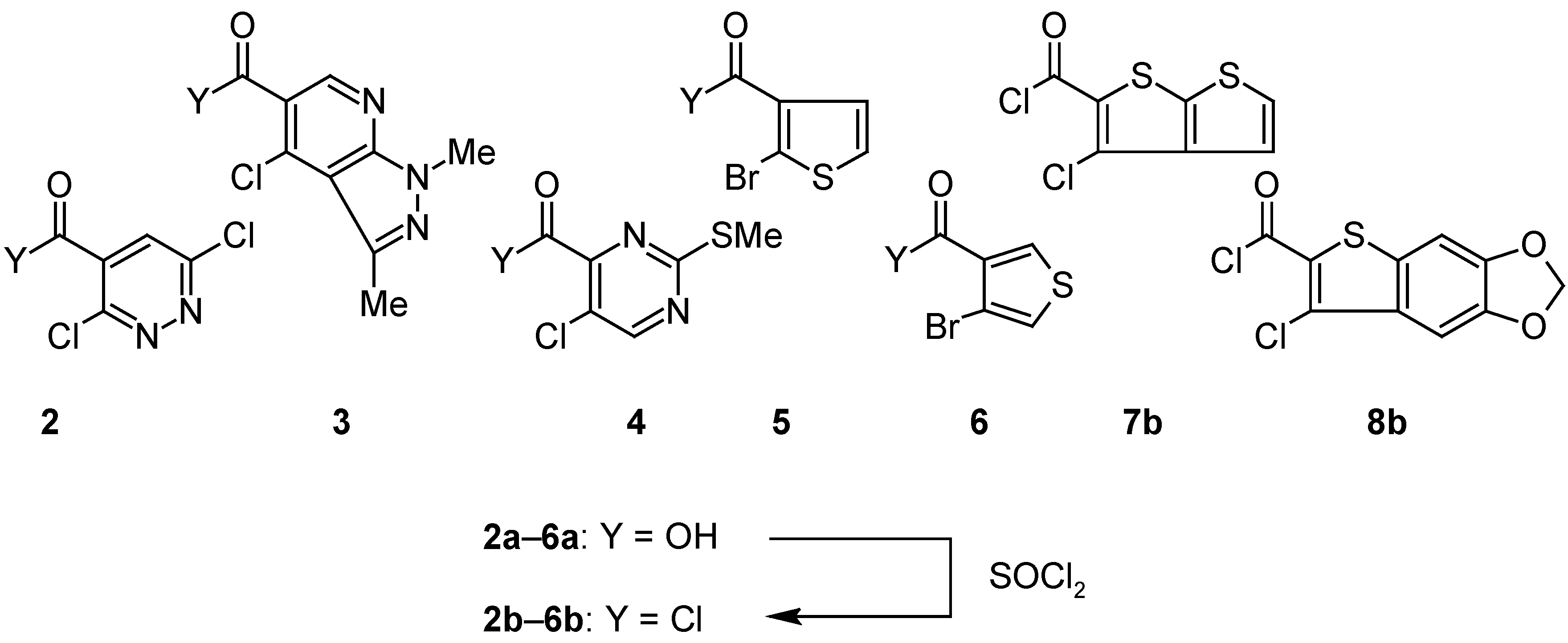
o-Haloarenecarbonyl Chlorides 2b–6b: General Procedure.
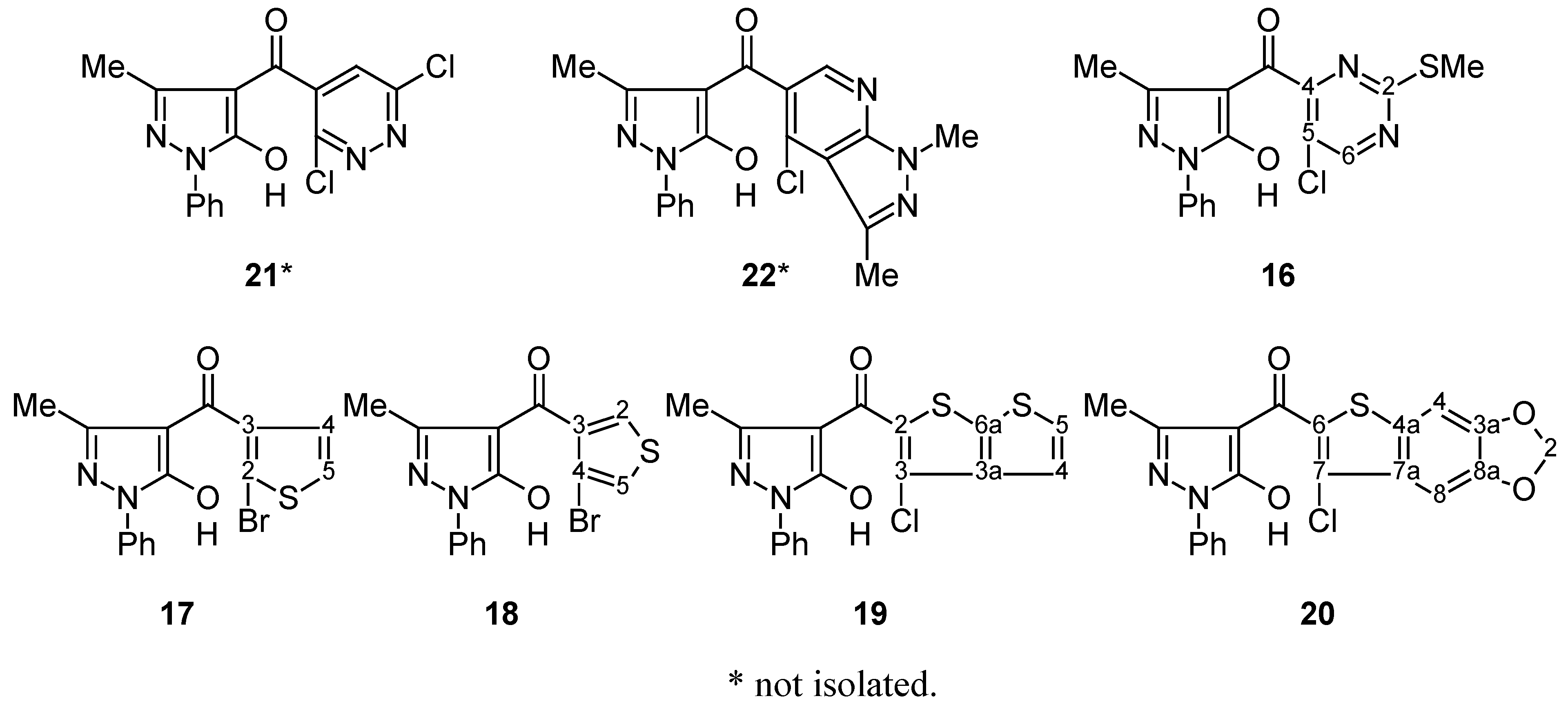
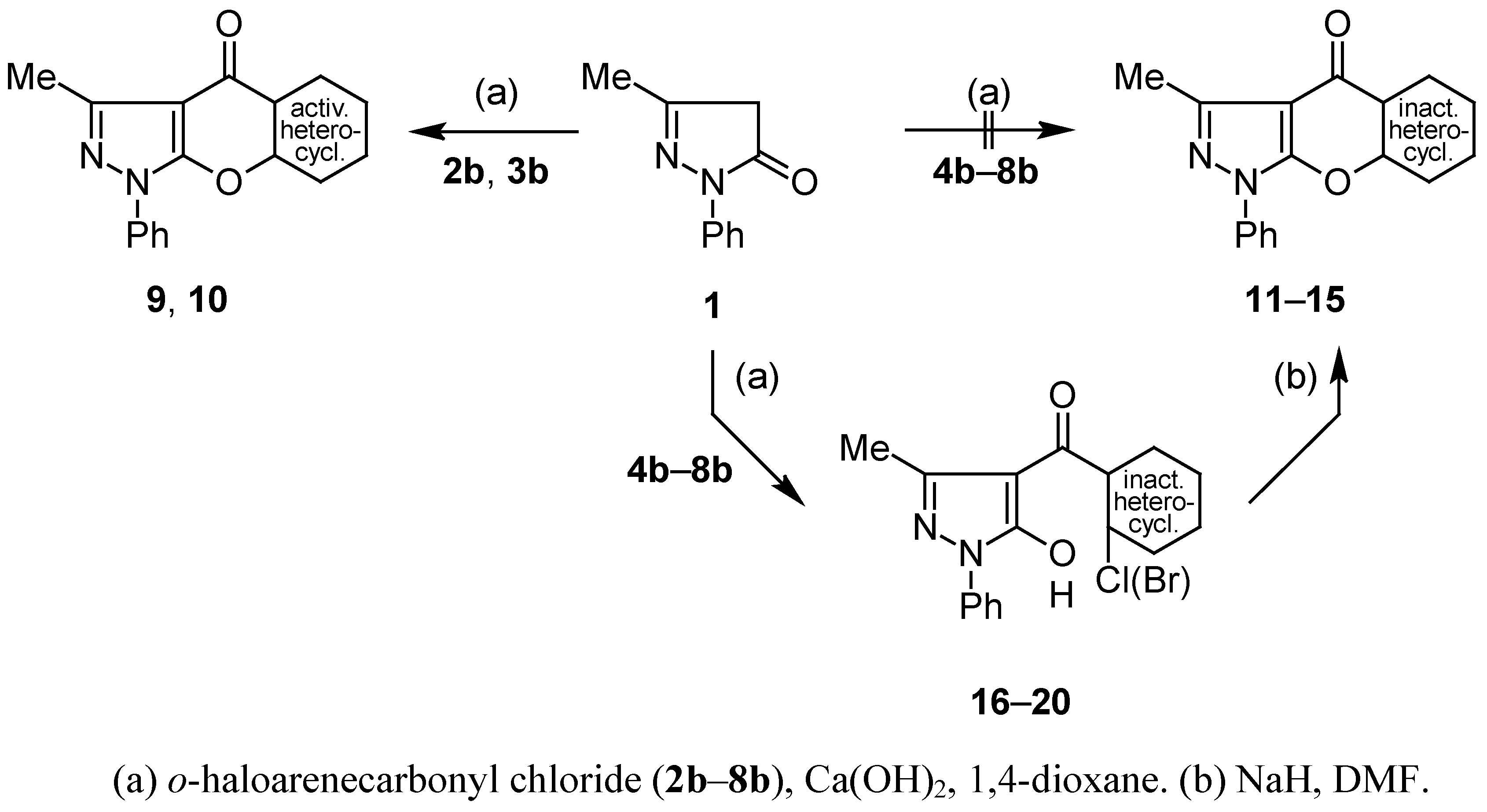
Compounds 9, 10, 16–20: General Procedure.
Cyclisation of 4-Aroylpyrazol-5-ols 16–20 into Compounds 11–15; General Procedure.
Acknowledgements
References
- Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. [Google Scholar] [CrossRef]
- Abraham, D. J., ed. Burger's Medicinal Chemistry and Drug Discovery, 6th ed.; Wiley-Interscience: New York, 2003; [Chem. Abstr. 2005, 143, 109740]. [Google Scholar]
- Makriyannis, A.; Biegel, D. Drug Discovery Strategies and Methods; Marcel Dekker, Inc.: New York, 2004; [Chem. Abstr. 2004, 140, 229394]. [Google Scholar]
- Eller, G. A.; Wimmer, V.; Haring, A. W.; Holzer, W. An Efficient Approach to Heterocyclic Analogues of Xanthone: A Short Synthesis of all possible Pyrido[5,6]pyrano[2,3-c]pyrazol-4(1H)-ones. Synthesis 2006, 4219–4229. [Google Scholar]
- Eller, G. A.; Haring, A. W.; Datterl, B.; Zwettler, M.; Holzer, W. Tri- and Tetracyclic Heteroaromatic Systems: Synthesis of Novel Benzo-, Benzothieno- and Thieno-Fused Pyrano[2,3-c]pyrazol-4(1H)-ones. Heterocycles 2007, 71, 87–104. [Google Scholar]
- Eller, G. A.; Datterl, B.; Holzer, W. manuscript submitted.
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed.; Thieme: Stuttgart, 2001; p. 99. [Google Scholar]
- Bell, J. Amlexanox for the Treatment of Recurrent Aphthous Ulcers. Clin. Drug Invest. 2005, 25, 555–566. [Google Scholar] [CrossRef]
- Colotta, V.; Cecchi, L.; Catarzi, D.; Melani, F.; Filacchioni, G.; Martini, C.; Tacchi, P.; Lucacchini, A. 1-(3-Aminophenyl)-3-methyl[1]benzopyrano[2,3-c]pyrazol-4-one: a new selective A2 adenosine receptor antagonist. Pharm. Pharmacol. Lett. 1992, 2, 74–76, [Chem. Abstr. 1993, 118, 32807]. [Google Scholar]
- Eller, G. A.; Holzer, W. The 4-Methoxybenzyl (PMB) Function as a Versatile Protecting Group in the Synthesis of N-Unsubstituted Pyrazolones. Heterocycles 2004, 63, 2537–2555. [Google Scholar] [CrossRef]
- Holzer, W.; Eller, G. A.; Wimmer, V.; Haring, A. manuscript in preparation.
- Minkin, V. I.; Garnovskii, A. D.; Elguero, J.; Katritzky, A. R.; Denisko, O. V. The tautomerism of heterocycles: Five-membered rings with two or more heteroatoms. Adv. Heterocycl. Chem. 2000, 76, 157–323, [Chem. Abstr. 2000, 133, 309576]. [Google Scholar] [CrossRef]
- Jensen, B. S. Synthesis of 1-phenyl-3-methyl-4-acyl-5-pyrazolones. Acta Chem. Scand. 1959, 13, 1668–1670, [Chem. Abstr. 1962, 56, 66890]. [Google Scholar] [CrossRef]
- Williams, A. Recent advances in NMR prediction and automated structure elucidation software. Curr. Opin. Drug Discovery Dev. 2000, 3, 298–305, [Chem. Abstr. 2000, 133, 222096]. [Google Scholar]
- NMRPredict. version 3.2.8; Modgraph Consultants, Ltd.: Herts, United Kingdom, 2004; www.modgraph.co.uk.
- ACD/C+H NMR Predictors and DB, version 10.04. Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2006; www.acdlabs.com.
- Braun, S.; Kalinowski, H.-O.; Berger, S. 150 and More Basic NMR Experiments: A Practical Course, 2nd ed.; Wiley-VCH: Weinheim, 1998; [Chem. Abstr. 1999, 131, 184497]. [Google Scholar]
- Neuhaus, D.; Williamson, M. P. The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd ed.; Wiley-VCH, Inc.: New York, 2000; [Chem. Abstr. 2004, 142, 392075]. [Google Scholar]
- Kover, K. E.; Batta, G. Theoretical and practical aspects of one- and two-dimensional heteronuclear overhauser experiments and selective 13C T1-determinations of heteronuclear distances. Prog. Nucl. Magn. Reson. Spectrosc. 1987, 19, 223–266, [Chem. Abstr. 1987, 107, 145869]. [Google Scholar] [CrossRef]
- Szántay, C, Jr.; Csepregi, Z.; Aranyosi, P.; Ruznák, I.; Töke, L.; Víg, A. Nuclear Magnetic Resonance Investigations of the Azo-Hydrazone Tautomerism of Azoreactive Dye Chromophores. Magn. Reson. Chem. 1997, 35, 306–310, [Chem. Abstr. 1997, 126, 294555]. [Google Scholar] [CrossRef]
- Davis, D. G.; Bax, A. Simplification of proton NMR spectra by selective excitation of experimental subspectra. J. Am. Chem. Soc. 1985, 107, 7197–7198. [Google Scholar]
- Sarkar, S. K.; Bax, A. A simple and sensitive one-dimensional NMR technique for correlation of proton and carbon chemical shifts. J. Magn. Reson. 1985, 62, 109–112. [Google Scholar]
- Bax, A. Structure determination and spectral assignment by pulsed polarization transfer via long-range proton-carbon-13 couplings. J. Magn. Reson. 1984, 57, 314–318. [Google Scholar]
- Jippo, T.; Kamo, O.; Nagayama, N. Determination of long-range proton-carbon 13 coupling constants with selective two-dimensional INEPT. J. Magn. Reson. 1986, 66, 344–348. [Google Scholar]
- Willker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar]
- ACD/Name, version 10.01. Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2006; www.acdlabs.com.
- Eller, G. A. Improving the Quality of Published Chemical Names with Nomenclature Software. Molecules 2006, 11, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for non-commercial purposes.
Share and Cite
Eller, G.; Holzer, W. A Convenient Approach to Heterocyclic Building Blocks: Synthesis of Novel Ring Systems Containing a [5,6]Pyrano[2,3-c]pyrazol-4(1H)-one Moiety. Molecules 2007, 12, 60-73. https://doi.org/10.3390/12010060
Eller G, Holzer W. A Convenient Approach to Heterocyclic Building Blocks: Synthesis of Novel Ring Systems Containing a [5,6]Pyrano[2,3-c]pyrazol-4(1H)-one Moiety. Molecules. 2007; 12(1):60-73. https://doi.org/10.3390/12010060
Chicago/Turabian StyleEller, Gernot, and Wolfgang Holzer. 2007. "A Convenient Approach to Heterocyclic Building Blocks: Synthesis of Novel Ring Systems Containing a [5,6]Pyrano[2,3-c]pyrazol-4(1H)-one Moiety" Molecules 12, no. 1: 60-73. https://doi.org/10.3390/12010060