Relative Stereochemistry of a Diterpene from Salvia cinnabarina

Abstract
:Introduction
Results and Discussion
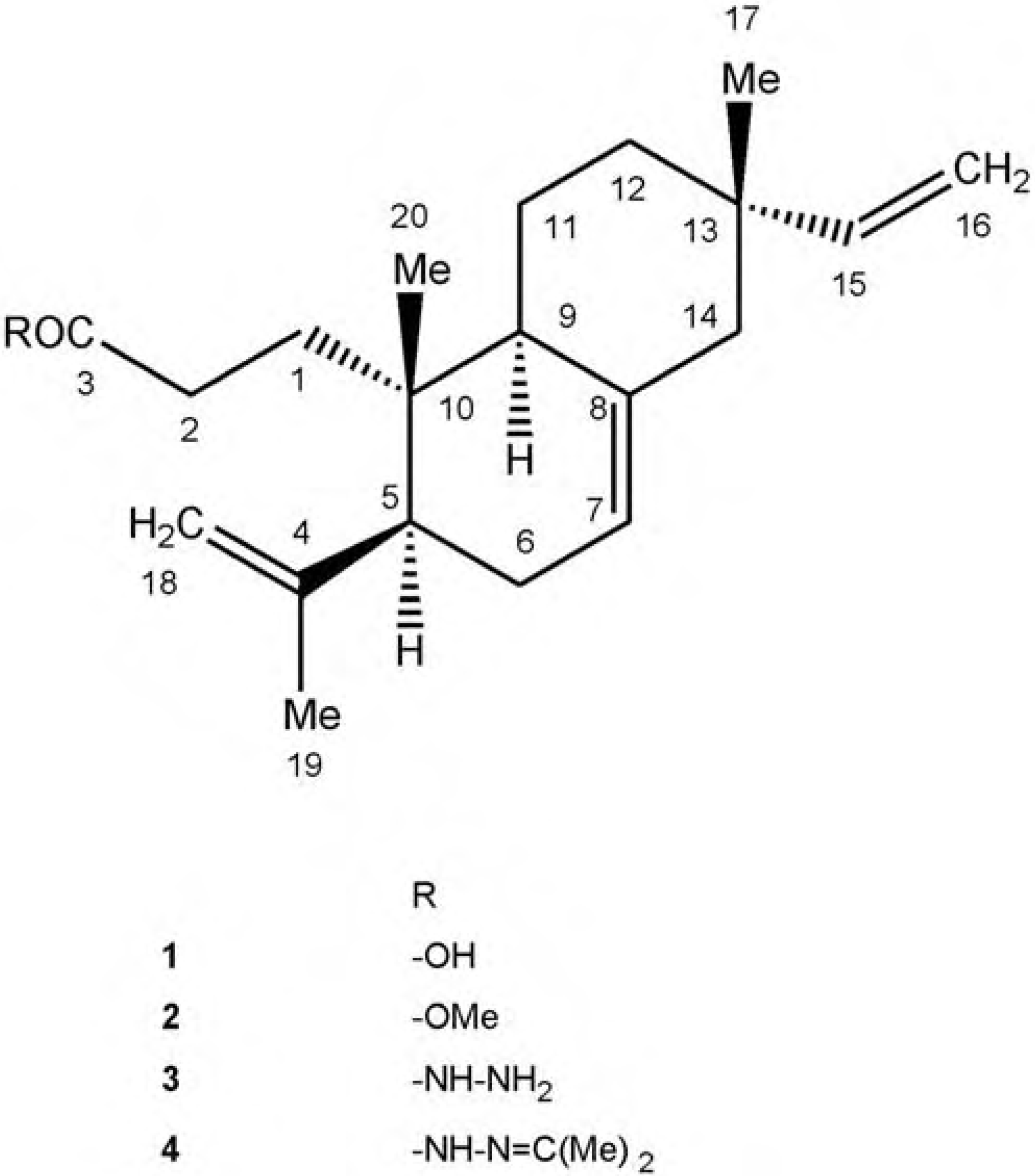

{kind=link}
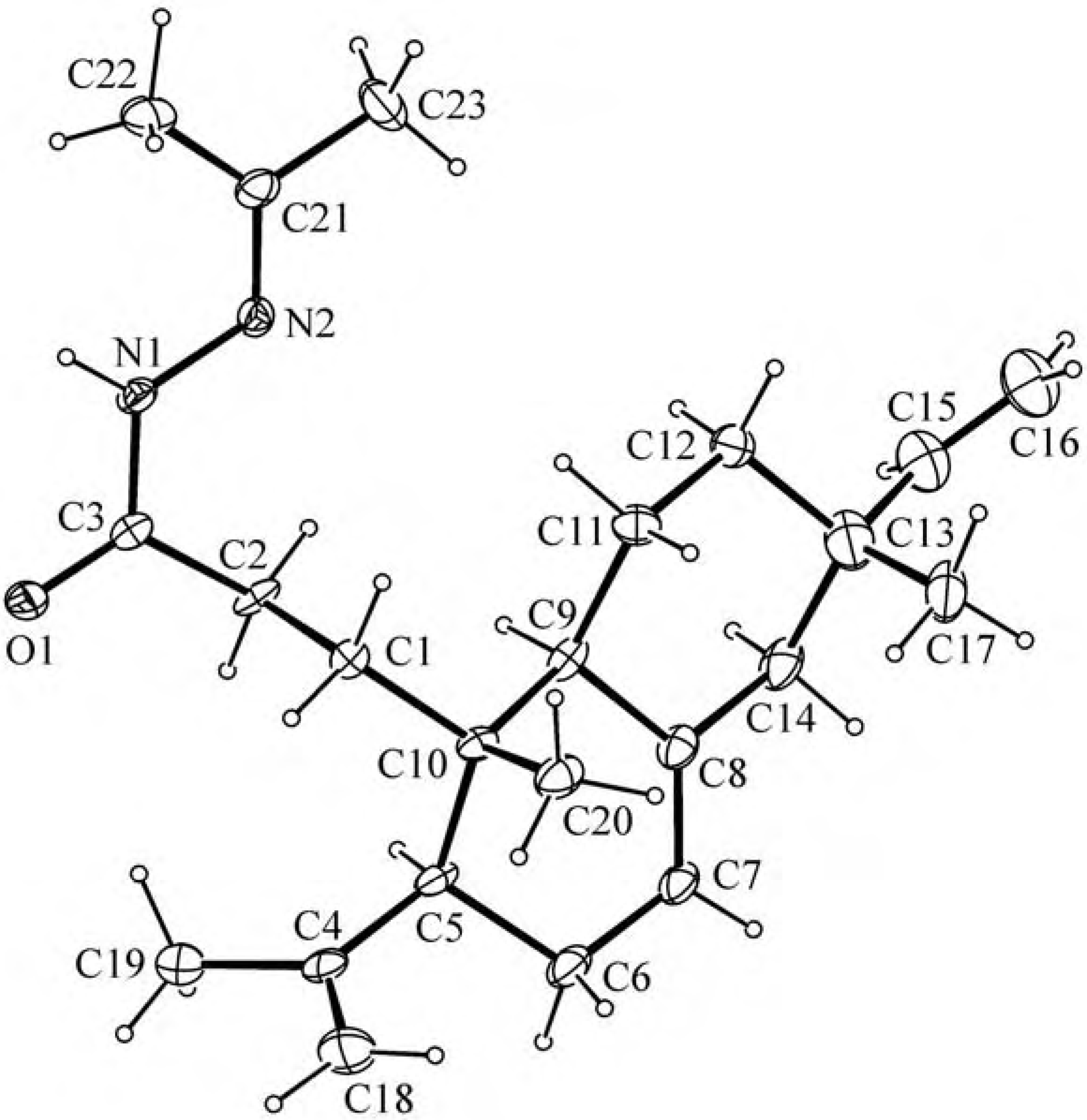
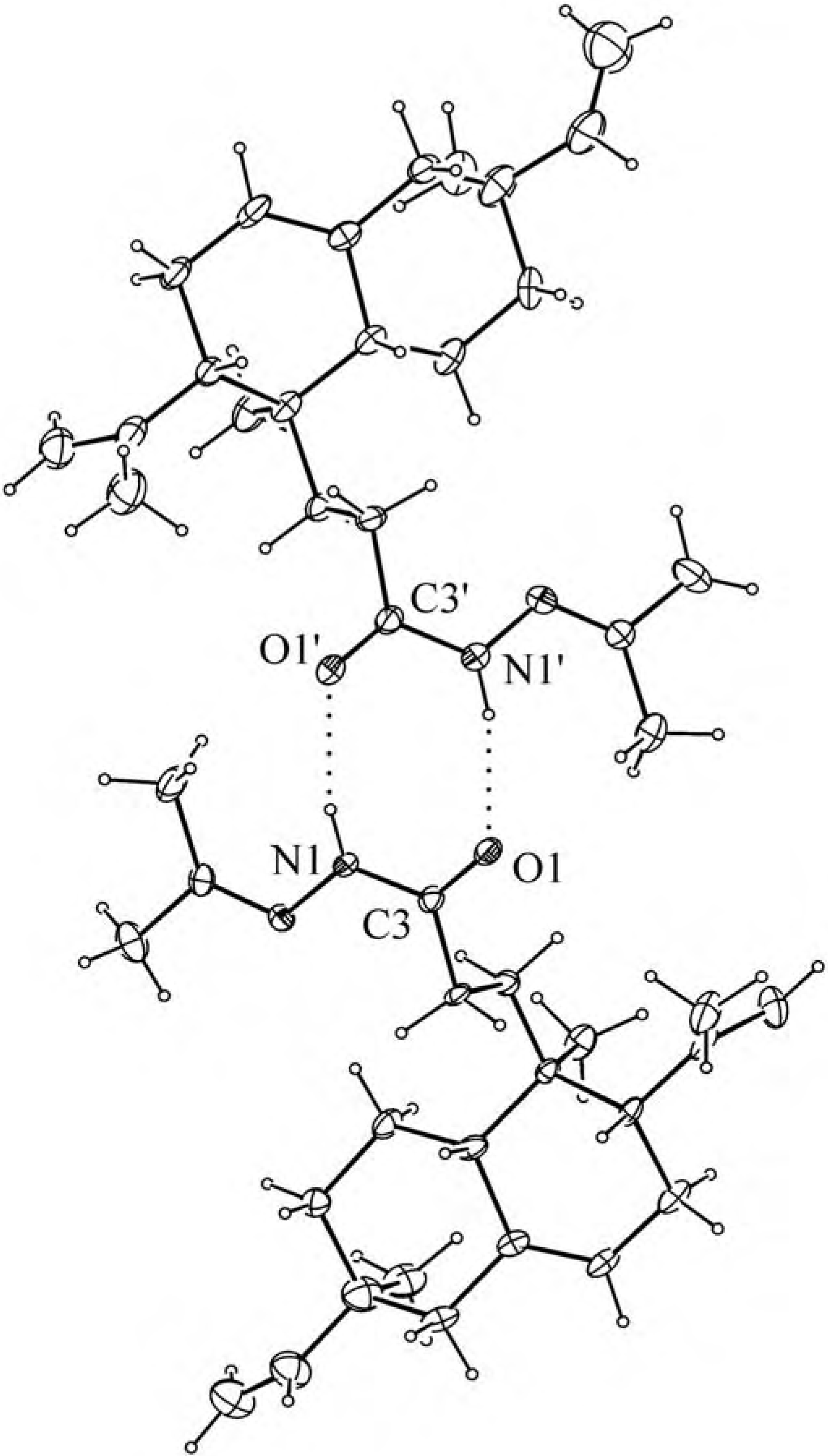{kind=link}
{kind=link}
| C | 1a | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 32.0 | 32.2 | 33.0 | 31.9 |
| 2 | 29.0 | 28.8 | 29.1 | 29.4 |
| 3 | 181.0 | 174.7 | 174.6 | 176.3b |
| 4 | 147.3 | 147.3 | 147.8 | 147.5 |
| 5 | 49.5 | 49.4 | 49.8 | 49.0 |
| 6 | 29.3 | 29.3 | 29.1 | 29.4 |
| 7 | 121.4 | 121.4 | 121.3 | 121.3 |
| 8 | 136.0 | 136.0 | 136.0 | 136.2 |
| 9 | 44.4 | 44.2 | 44.2 | 43.9 |
| 10 | 37.6 | 37.6 | 37.5 | 37.8 |
| 11 | 21.0 | 20.9 | 20.8 | 20.8 |
| 12 | 36.3 | 36.3 | 36.2 | 36.3 |
| 13 | 37.0 | 37.0 | 36.9 | 37.0 |
| 14 | 46.4 | 46.4 | 46.4 | 46.4 |
| 15 | 150.1 | 150.1 | 150.0 | 150.3 |
| 16 | 109.4 | 109.4 | 109.4 | 109.3 |
| 17 | 21.5 | 21.5 | 21.5 | 21.5 |
| 18 | 114.0 | 113.9 | 113.9 | 113.7 |
| 19 | 23.7 | 23.7 | 23.3 | 23.9 |
| 20 | 16.7 | 16.7 | 16.8 | 17.0 |
| COOMe | 51.5 | |||
| C=N | 148.6c | |||
| Acetone | ||||
| moiety Me | 25.4 and 27.2 |
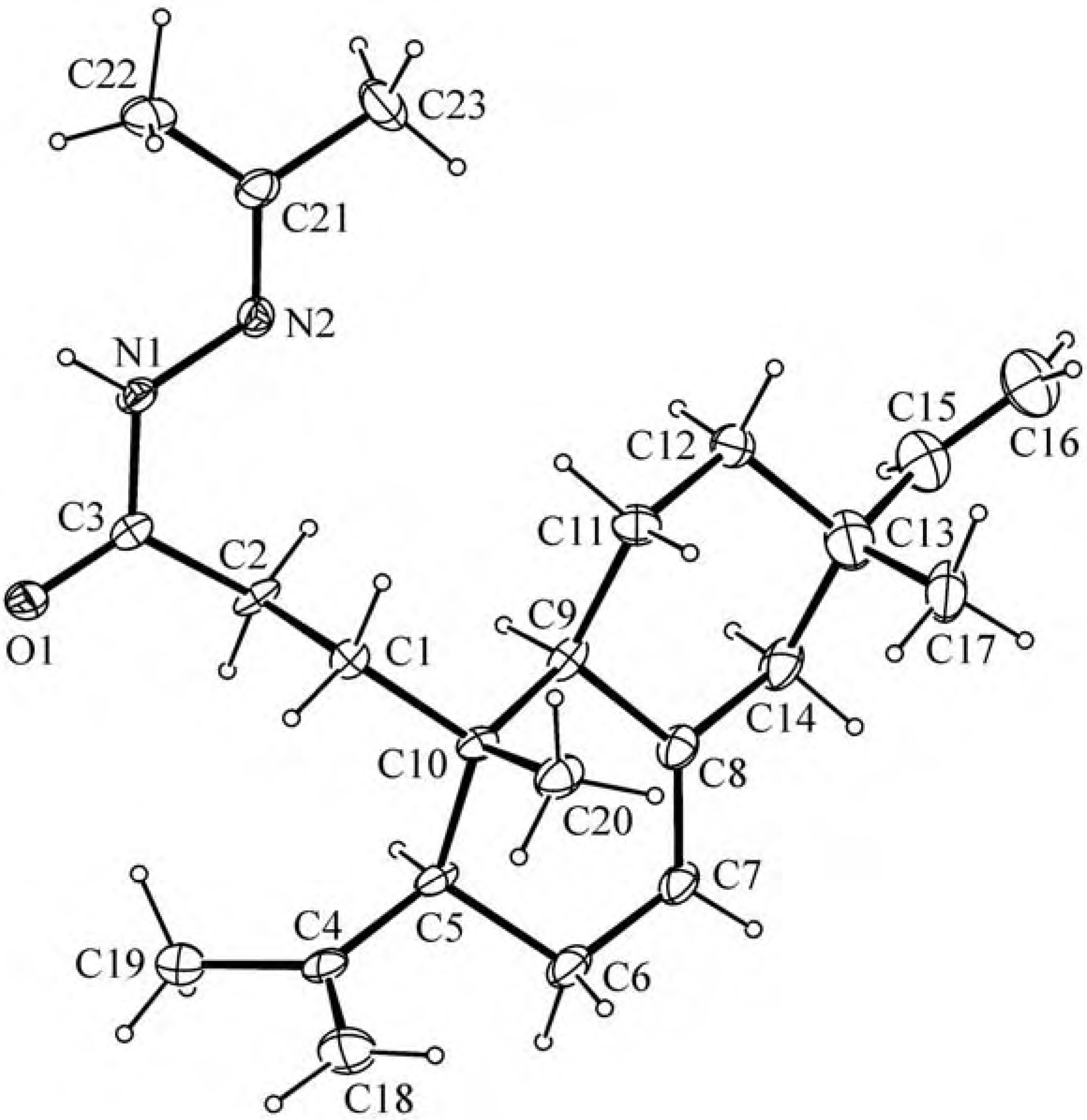
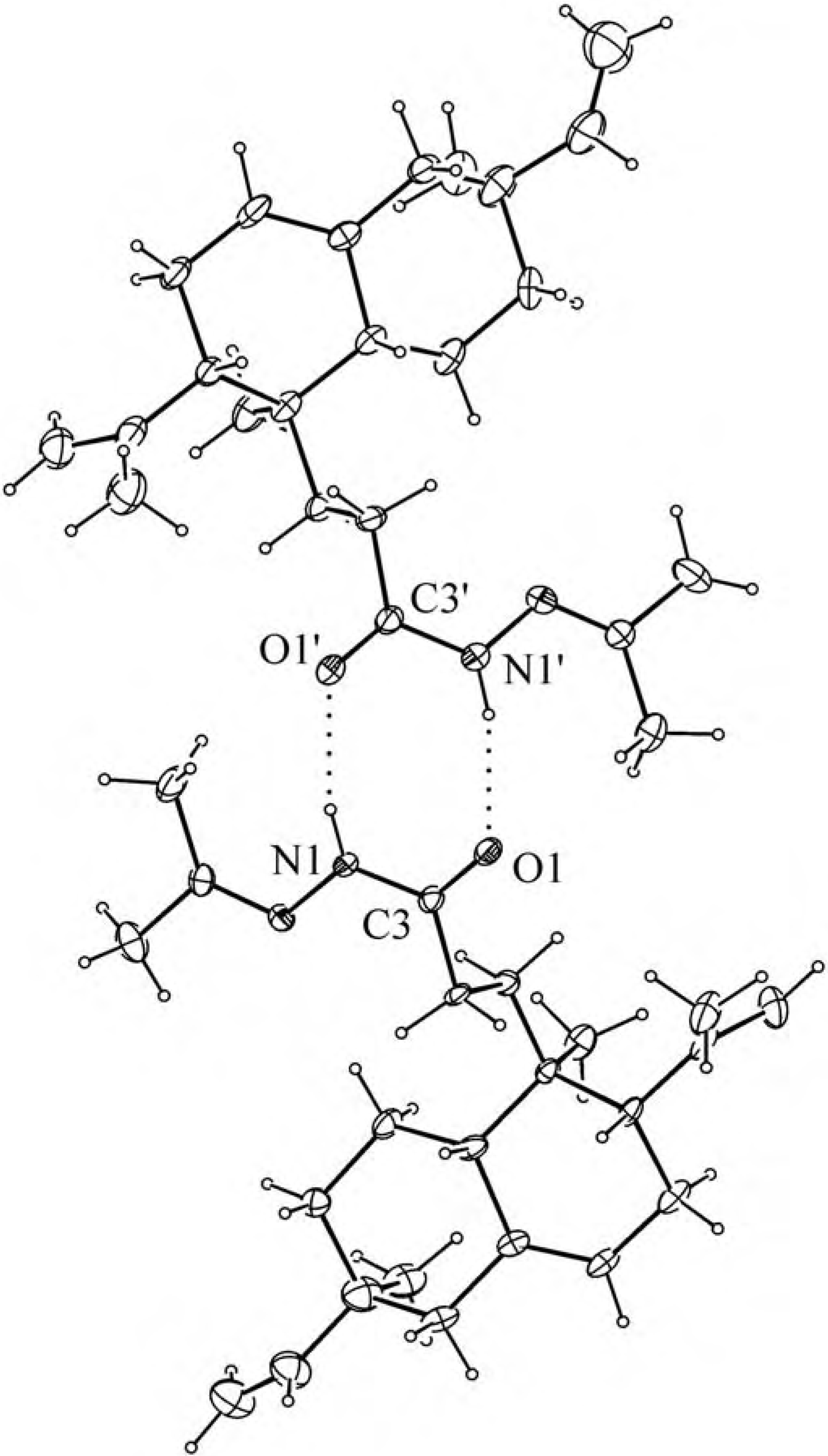
Experimental
General
Extraction
Synthetic procedures
X-ray structure determination of 4
Acknowledgments
References and Notes
- Penso, G. Index Plantarum Medicinalium Totius Mundi Eorumque Synonymorum; O.E.M.F.: Milano, 1983; pp. 845–848. [Google Scholar]
- Tomás-Barberán, F.A.; Wollenweber, E. Flavonoid aglycones from the leaf surfaces of some Labiatae species. Plant Syst. Evol. 1990, 173, 109–118. [Google Scholar] [CrossRef]
- Rodríguez-Hahn, L.; Esquivel, B.; Cárdenas, J. Clerodane Diterpenes in Labiatae. In Progress in the Chemistry of Organic Natural Products; Zechmeister, L., Ed.; Springer: Heidelberg, 1994; Vol. 63, 162 and literature cited therein. [Google Scholar]
- Cuvelier, M.E.; Richard, H.; Berset, C. Antioxidative Activity and Phenolic Composition of Pilot- Plant and Commercial Extracts of Sage and Rosemary. J. Am. Oil Chem. Soc. 1996, 73, 645–652. [Google Scholar] [CrossRef]
- Romussi, G.; Ciarallo, G.; Bisio, A.; Fontana, N.; De Simone, F.; De Tommasi, N.; Mascolo, N.; Pinto, L. A new diterpenoid with antispasmodic activity from Salvia cinnabarina. Planta Med. 2001, 67, 153–155. [Google Scholar]
- Capasso, R.; Izzo, A.A.; Romussi, G.; Capasso, F.; De Tommasi, N.; Bisio, A.; Mascolo, N. A diterpenoid from Salvia cinnabarina inhibits rat urinary bladder contractility in vitro. Planta Med. 2004, 70, 185–188. [Google Scholar]
- Capasso, R.; Izzo, A.A.; Capasso, F.; Romussi, G.; Bisio, A.; Mascolo, N. A diterpenoid from Salvia cinnabarina inhibits mouse intestinal motility in vivo. Planta Med. 2004, 70, 375–377. [Google Scholar]
- Capasso, R.; Avello, G.; Ascione, V.; D’Alessio, A.; De Fusco, R.; Romussi, G.; et al. Antimicrobial activity of a secoisopimarane diterpenoid isolated from Salvia cinnabarina. In Proceedings 3rd International Symposium on Natural Drugs, Naples; 2003; pp. 193–197. [Google Scholar]
- Alfieri, A.; Maione, F.; Bisio, A.; Romussi, G.; Mascolo, N.; Cicala, C. Effect of a diterpenoid from Salvia cinnabarina on arterial blood pressure in rats. Phytother Res. 2007, 21, 690–692. [Google Scholar]
- Duisenberg, A.J.M. Indexing in single-crystal diffractometry with an obstinate list of reflections. J. Appl. Cryst. 1992, 25, 92–96. [Google Scholar] [CrossRef]
- Hooft, R.W.W. COLLECT; Nonius BV: Delft, The Netherlands, 1999. [Google Scholar]
- Sheldrick, G.M. SADABS. Version 2.10; University of Göttingen: Göttingen, Germany, 2003. [Google Scholar]
- Altomare, A.; Burla, M.C.; Cavalli, M.; Cascarano, G.L.; Giacovazzo, C.; Gagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Kato, T.; Kabuto, C.; Sasaki, N.; Tsunagawa, M.; Aizawa, H.; Fujita, K.; Kato, Y.; Kitahara, Y.; Takahashi, N. Momilactones, growth inhibitors from rice, Oryza sativa. Tetrahedron Lett. 1973, 39, 3861–3864. [Google Scholar]
- Lipson, V.V.; Desenko, S.M.; Orlov, V.D.; Shishkin, O.V.; Shirobokova, M.G.; Chernenko, V.N.; Zinov’eva, L.I. Cyclocondensation of 3-amino-1,2,4-triazoles with esters of substituted cinnamic acids and aromatic unsaturated ketones. Chem. Heterocycl. Comp. 2000, 36, 1329–1335. [Google Scholar]
- Khusainova, N.G.; Mostovaya, O.A.; Azancheev, N.M.; Litvinov, I.A.; Krivolapov, D.B.; Cherkasov, R.A. Reaction of 2-acetyl-5-methyl-2H-1,2,3-diazaphosphole with butane-2,3-diol. Mendeleev Commun. 2004, 5, 212–214. [Google Scholar]
- Atta-Ur-Rahman; Akhtar, M.N.; Choudhary, M.I.; Tsuda, Y.; Yasin, A.; Sener, B.; Parvez, M. New diterpene isopimara-7,15-dien-19-oic acid and its prolyl endopeptidase inhibitory activity. Nat. Prod. Res. 2005, 19, 13–22. [Google Scholar]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1 - 4 are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Bisio, A.; Pagano, B.; Romussi, A.; Bruno, O.; De Tommasi, N.; Romussi, G.; Mattia, C.A. Relative Stereochemistry of a Diterpene from Salvia cinnabarina. Molecules 2007, 12, 2279-2287. https://doi.org/10.3390/12102279
Bisio A, Pagano B, Romussi A, Bruno O, De Tommasi N, Romussi G, Mattia CA. Relative Stereochemistry of a Diterpene from Salvia cinnabarina. Molecules. 2007; 12(10):2279-2287. https://doi.org/10.3390/12102279
Chicago/Turabian StyleBisio, Angela, Bruno Pagano, Alessia Romussi, Olga Bruno, Nunziatina De Tommasi, Giovanni Romussi, and Carlo Andrea Mattia. 2007. "Relative Stereochemistry of a Diterpene from Salvia cinnabarina" Molecules 12, no. 10: 2279-2287. https://doi.org/10.3390/12102279